- The three actions concern EP (UK) 1 893 196 B2 (the “Patent”) and SPC/GB13/079 (the “SPC”). The Patent and SPC are owned by the First Defendant and it is accepted for the purposes of these actions that the Second Defendant is the exclusive licensee of both the Patent and the SPC. There is no need to distinguish between the Defendants and I shall refer to them simply as ‘Astellas’.
- In the three actions the Accord parties, the Sandoz parties and the Teva parties (collectively ‘the Claimants’) seek to revoke the Patent and invalidate the SPC. If the Patent (and SPC) are valid, the Claimants have each confirmed there is no issue on infringement. The relevant date is the application date, 29 March 2006 (‘the Application Date’).
- The Patent claims the compound enzalutamide. Enzalutamide arose from a research and development project undertaken by the University of California in the years leading up to 2006, to try to find a better treatment for prostate cancer. Astellas assert that enzalutamide has proved to be spectacularly successful, but there is no plea of commercial success. It is marketed by Astellas under the brand name “Xtandi”. Astellas say that Xtandi’s success has been marked not just against hormone sensitive prostate cancer (“HS prostate cancer or HSPC”) but also against the far more problematic hormone refractory (or castrate resistant) prostate cancer (“HR prostate cancer or HRPC”). The Patent is the primary patent, disclosing and claiming the compound per se in addition to its therapeutic use.
- The Claimants rely on two pieces of prior art, both originating from the inventors discussing their own work. The first is a series of slides presented as part of a lecture given by an inventor, Dr Sawyers. These are referred to as the “Slides”. The second is referred to as the “Poster”. The Slides and the Poster were presented at the same conference in September/October 2005. Each one is relied upon as a documentary disclosure. There is no dispute that the Slides and the Poster were made available to the public in so far as these actions are concerned.
- It is the Claimants’ case that the Slides and the Poster each render enzalutamide (and its use in prostate cancer therapy) obvious. Alternatively, the Claimants say that the Patent does not plausibly disclose a technical contribution at all. The Claimants also seek to exert an insufficiency squeeze in the event that the Patent is not obvious.
- Each piece of prior art discloses a molecule identified as RD162 which, as I understand matters, is agreed to be the closest prior art molecule. The compound claimed in claim 1 is identified as RD162'. The structures of these compounds are as follows:
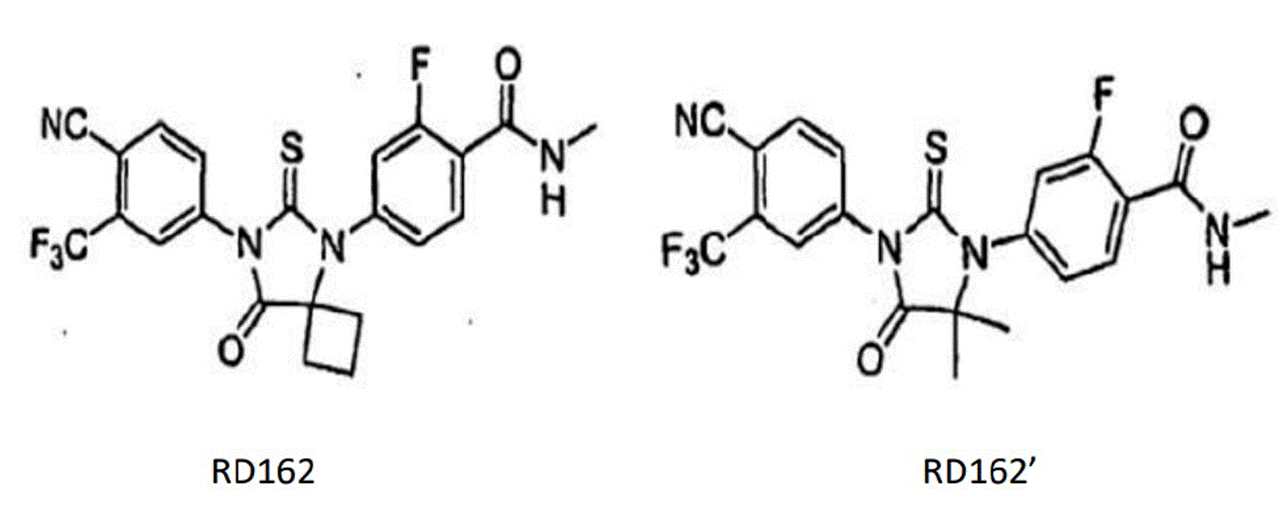
- In these molecules, the only difference lies in the substituents at the bottom right of the central thiohydantoin ring at what I will call position X (to avoid confusion with the pharmacophore on the Slides): where RD162 has a cyclobutyl group and RD162' has a geminal dimethyl substituent (i.e. two methyl groups on the same carbon).
- Although the comparison I have set out above embodies 20:20 hindsight and, for that reason, is exactly the opposite of what is required for a resolution of the obviousness arguments, it nonetheless explains how and why most of the issues arise.
- It is necessary to analyse the obviousness case which the Claimants put forward and various reasons advanced by Astellas as to why RD162' was inventive.
- The parties agreed a technical primer on the clinical treatment of prostate cancer together with an agreed statement of common general knowledge. I am grateful to the parties for agreeing these documents. Only one point was identified as in dispute as to the CGK and I address that below.
- There were no fact witnesses, but both sides have submitted evidence under CEA notices. Most of the material relied upon in the CEA Notices is no longer relevant, in view of agreements made in these actions only as to the Slides and the Poster. To the extent that any of the material remains relevant, I refer to it below.
- Each side called two expert witnesses:
i) The cancer biologists were Prof Hickson for the Claimants and Prof Clarke for Astellas.
ii) The medicinal chemists were Prof Westwell for the Claimants and Prof Ward for Astellas.
- Prof Hickson is a Professor of Cancer Drug Discovery Bioscience at the Translational and Clinical Research Institute, Newcastle University Faculty of Medical Sciences.
- As the Claimants submitted, Prof Hickson has a wealth of experience in designing and performing suitable in vitro and in vivo assays for assessing the potential of inhibitors and specifically screening compounds for antagonism and agonism of AR activity with PSA, ELISA, western blot and luciferase reporter assays. Astellas made no criticism of Prof Hickson.
- Prof Clarke is a consultant urologist (also known as a urological surgeon), with a special interest in urological oncology, at the Christie and Salford Royal Hospitals in Manchester. Between 2003 - 2009 (encompassing the Application Date), he was Chairman of the UK National Cancer Research Institute Prostate Clinical Studies Group and integrally involved in the development of a number of clinical trials in prostate cancer. He specialises in prostate, bladder, renal and testis cancers, both in respect of research into these genito-urinary malignancies and the diagnosis and delivery of cancer treatment of these conditions in healthcare systems. His research interests encompass basic/translational science and clinical trials in urological cancer.
- In their closing, the Claimants acknowledged that Prof Clarke is an eminent consultant urologist but they contended he was the wrong witness for Astellas to call. Their criticism related to a rather narrow point, as I understood matters. Although the Claimants acknowledged he is an eminent clinician, they suggested he has never been a cancer biologist skilled in testing, in a preclinical setting, the relative performance of candidate drugs in vivo and in vitro - the topics on which he was asked to give evidence. Whether the Claimants’ criticisms have any merit on this topic is one best considered in the relevant context, but, as will be seen at [418] below, these criticisms came to nothing.
- It emerged that Prof Clarke wrote the technical primer and for that I am extremely grateful.
- Prof Westwell is a Professor of Medicinal Chemistry at the School of Pharmacy and Pharmaceutical Sciences at Cardiff University. Between 1996 - 2006, Prof Westwell worked as a senior medicinal chemistry research fellow within the Cancer Research Laboratories at the University of Nottingham which focussed on small molecule cancer drug discovery.
- As the Claimants submitted, Prof Westwell had personal experience of drug design based on the AR as the drug target including the in vitro and in vivo profiling of new drug candidate molecules as compared with the standard of care (bicalutamide). Astellas made no personal criticism of Prof Westwell as a witness. Their major point was that hindsight had infected his evidence. This is a point I must consider in greater detail later.
- Prof Ward is the Sêr Cymru Professor in Translational Drug Discovery, and Director of the Medicines Discovery Institute, at Cardiff University. From 2001 to 2010, he was Associate Director at GlaxoSmithKline (GSK) where he led medicinal chemistry groups and multidisciplinary project teams (mainly consisting of research biologists and chemists) through all stages of drug development from target identification to early clinical studies. He has c.30 years’ experience in drug discovery and development, leading to the identification of more than 15 clinical candidates across the biotech, pharma, and academia environments including with specialist experience in drugs against cancer.
- The Claimants were highly critical of Prof Ward, devoting a whole Annex in their Closing to that topic (which elicited a full response from Astellas). All these points are bound up in the obviousness arguments and are best addressed in that context. Anticipating my conclusions below, some of the Claimants’ criticisms were justified but the attack went over the top.
- Notwithstanding the criticisms levelled by the parties, I am grateful to all four experts for their contributions to the evidence, which facilitated the agreed technical primer and the very largely agreed statement of the CGK. All four provided me with the technical education the Court required to put itself into the position of the Skilled Team at the priority date.
- There was no dispute about the skillset of the Skilled Team. The team would be interested in seeking to develop a new drug for the treatment of prostate cancer and, in view of the Claimants’ reliance on the Slides and the Poster, in particular they would be interested in developing a new antagonist to the androgen receptor. The team would include a person with relevant knowledge and skills in the relevant biology of prostate cancer (‘the Skilled Cancer Biologist’) and a person with knowledge and skills in medicinal chemistry (‘the Skilled Medicinal Chemist’).
- The Skilled Cancer Biologist would understand the underlying disease and biological target and use that understanding to develop a hypothesis for how the disease may be treated. The Skilled Cancer Biologist would select and/or devise suitable in vitro and in vivo testing.
- The Skilled Medicinal Chemist would have an undergraduate degree in chemistry, most likely a PhD, and experience in industry. He or she is well-versed in drug design, synthesis, optimisation, purification and characterisation. The skilled chemist would interpret in vitro and in vivo data for any novel therapy candidates that may be created during the research process and evaluate a compound’s physicochemical properties, pharmacokinetics and wider drug properties, which work may be done in conjunction with the Skilled Cancer Biologist.
- Prof Westwell discussed potential differences between chemists in industry and academia (saying that the latter are more focussed on publication of novel data rather than having a more commercial focus) - see Westwell 1 §3.11. Prof Ward’s view is that commercial factors play a part in both academia and industry (Ward 2 §10-13).
- Whatever might be the approach amongst some academics, there can be no dispute that the skilled person of patent law does not just make compounds for compounds’ sake. As the TBA of the EPO in T 939/92 AgrEvo/Triazoles made clear (at [2.4.2]), “the notional “person skilled in the art” is not to be assumed to seek to perform a particular act without some concrete technical reason: he must, rather, be assumed to act not out of idle curiosity but with some specific technical purpose in mind”.
- What I set out in this section is based on the primer and the agreed statement of CGK. I address the one CGK point in dispute at the end.
- The prostate is a gland of the male reproductive system. The main purpose of the prostate is to produce fluid which mixes with sperm from the testes to form seminal fluid (semen). Semen includes enzymes like prostate-specific antigen (“PSA”), as well as other substances made by the seminal vesicles (the glands that compose semen) and prostate, such as zinc, citrate, and fructose.
- The prostate is located in front of the rectum and just below the bladder. It is about the size of a chestnut and is somewhat conical in shape. The prostate consists of a base, an apex, an anterior, a posterior and two lateral surfaces. The base is directed upward near the inferior surface of the bladder and the greater part of this surface is directly continuous with the bladder wall. The apex is directed downward and is in contact with the superior fascia of the urogenital diaphragm. See Figure 1 below.

Figure 1. Anatomy of the prostate gland.
- The prostate gland consists of four major zones based on histological features, shown in Figure 2 below:
i) Transition zone – glandular tissue constituting of two small lobules that surround the prostatic urethra;
ii) Central zone – glandular tissue surrounding the ejaculatory duct apparatus and which makes up the majority of the prostatic base;
iii) Peripheral zone – glandular tissue at the back of the prostate, which extends posterolaterally around the gland from the apex to the base and contains the majority of prostatic glandular tissue; and
iv) Anterior fibromuscular stroma – forms the entire anterior surface of the prostate as a thick, nonglandular layer, composed of vertically oriented smooth muscle bundles continuous with the bladder smooth muscle.
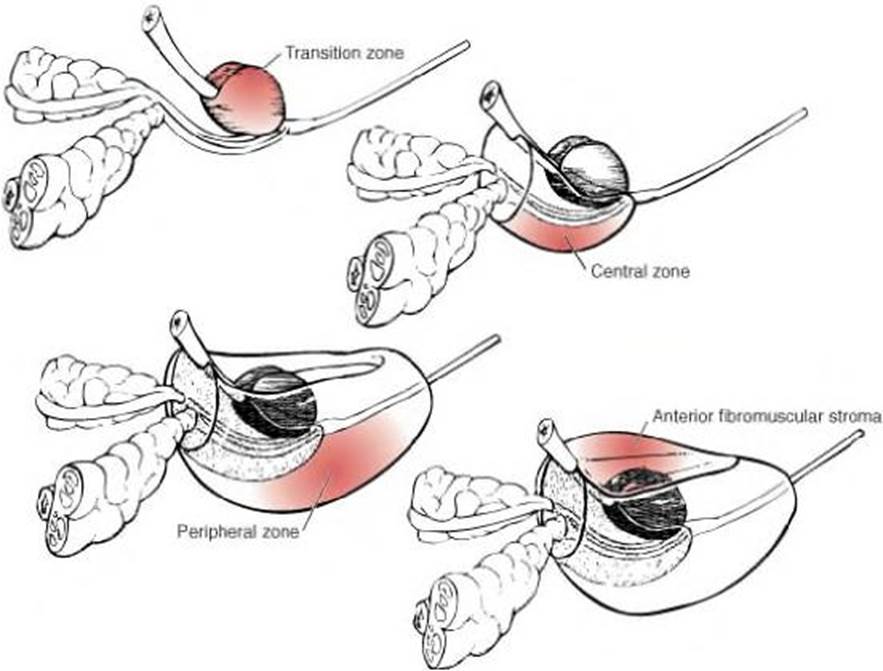
Figure 2. Normal prostate anatomy.
- The prostate is composed of branching glandular tissue, with ducts that are lined with secretory epithelial cells and basal cells, arranged in “acini” (clusters of cells that resemble a many-lobed berry). Secretory epithelial cells represent the major cell type in the gland. They are androgen-dependent for growth, and produce PSA and prostatic acid phosphatase. The basal cell layer is believed to contain the stem cell population for the epithelial prostate cells.
- The “prostatic stroma” is the supporting fibromuscular tissue of the prostate which separates the ducts from one another.
- The prostate is an androgen-dependent gland and requires androgens for normal function. Androgens are steroid hormones that are involved in prostate development and normal prostate function. The predominant and most active androgens in men are testosterone and its metabolite 5α-dihydrotestosterone (‘DHT’).
- Androgens bind to a protein called the androgen receptor (‘AR’). The AR is a steroid hormone receptor and is found in many of the body’s tissues, including in muscle, bone, brain, skin, and the genitourinary and gastrointestinal tracts. The AR functions as a ligand-inducible transcription factor, which means that, upon binding of a ligand (such as testosterone or DHT), the AR modulates the expression of certain genes.
- The AR’s natural ligands are the natural steroid hormones, testosterone and DHT. When testosterone or its active metabolite DHT bind to the AR, they activate (i.e. agonise) the receptor and thereby initiate a cascade of processes that contribute to the survival, growth, and proliferation of normal and cancerous prostate cells.
- Testosterone is synthesised mainly by the testes. Its synthesis is regulated by the action of androgens on the hypothalamic-pituitary-gonadal axis, a hormone-driven link between the brain and the testes. In the hypothalamus (which is in the front part of the brain), luteinising hormone-releasing hormone (‘LHRH’) is released and this interacts with LHRH receptors, stimulating the pituitary gland to release luteinising hormone (‘LH’) into the circulation. LH then binds to LH receptors in the testes, inducing the production of testosterone, which is synthesised through a series of biochemical steps from cholesterol. When testosterone levels rise above a certain threshold level, feedback inhibition (a physiological process known as a homeostatic “feedback loop”) reduces LHRH and LH production, thereby maintaining serum testosterone within carefully regulated physiological limits.
- Testosterone is transported via the blood stream to target tissues, where it enters cellular structures (including prostate epithelial cells) and is converted to DHT. DHT binds tightly to the AR in the cytoplasm of these cells. Prior to being bound to the androgen ligand, the AR resides in a dormant state in the cytoplasm of prostate cells, where it is bound to a “chaperone” protein known as a “heat-shock” protein (‘HSP’). This, and other co-chaperones, stabilise the structure. The AR undergoes a conformational change when the ligand, testosterone or DHT, binds to it. The AR then dissociates from its accompanying HSP, becomes phosphorylated and translocates from the cytoplasm to the nucleus. The AR forms homodimers (i.e. with itself), becoming an “AR-DHT complex”. The ligand-based activation of the AR is depicted below.
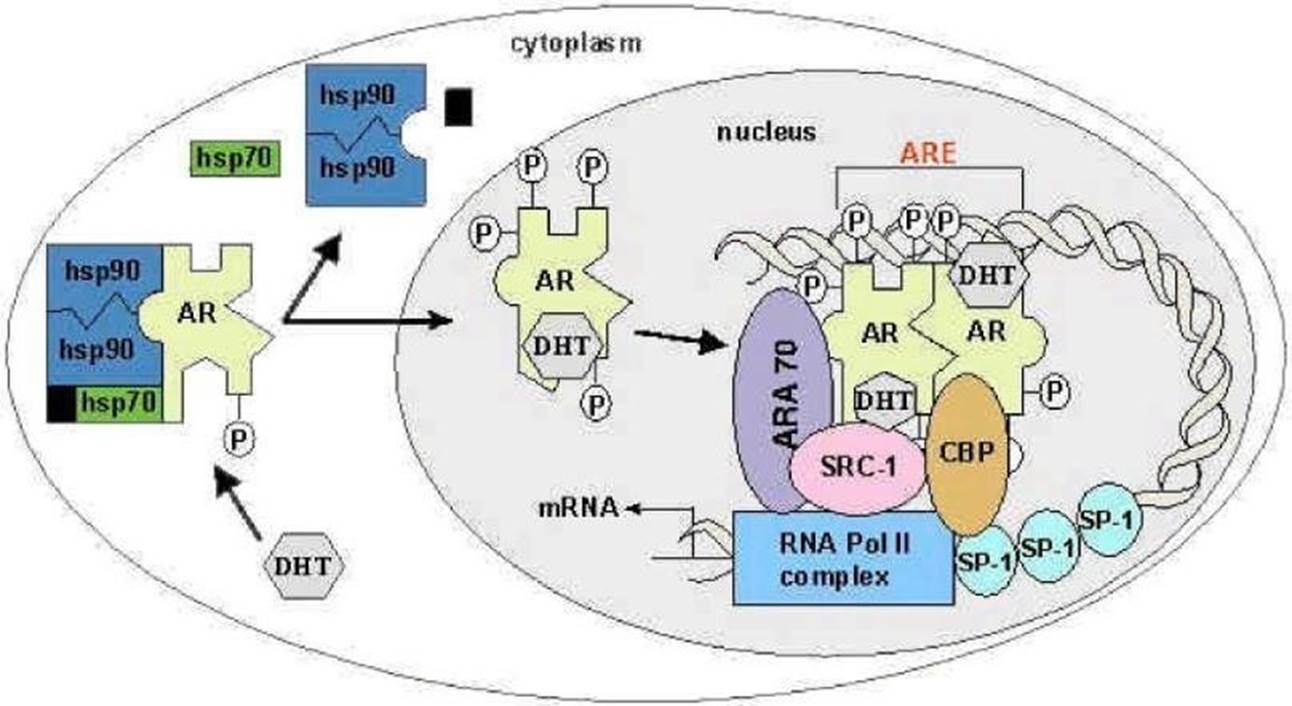
- Once in the nucleus AR activates transcription of genes that regulate cell growth and survival. AR dimers bind to DNA sequences known as androgen response elements (‘ARE’) in the regulatory region (the nuclear hormone binding site) of genes that are under a regulated transcription programme. Binding of AR dimers to ARE(s) in a regulatory region of an androgen responsive gene will cause the recruitment of transcription machinery. The AR-DHT complex recruits coactivators, co-regulatory proteins, and transcriptional machinery, such as RNA polymerase II, that aid in transcription (the first step of gene expression).
- One of the genes whose transcription the AR regulates is KLK3, which encodes for production of prostate specific antigen (‘PSA’). The PSA gene has an upstream promoter and enhancer region that the AR binds to. The genes that encode androgen dependent gene products are expressed first by transcription of the gene to messenger RNA (mRNA) and then translation of this message to protein.
- This androgen-induced transcriptional activity of AR is further modulated by the interaction of the AR with coregulators and by post-translational modification e.g. phosphorylation of the AR and the AR’s coregulators in response to other signalling cascades (for example insulin like growth factor, epidermal growth factor and cytokines such as IL-6).
- There are two main types of prostate cancer, prostatic adenocarcinoma and primary small cell carcinoma (although prostatic adenocarcinoma is the most common). Carcinoma is a cancer that forms in epithelial tissue. Epithelial tissue lines most organs, internal passages in the body, and the skin. In prostatic adenocarcinoma, the carcinoma forms in the gland cells that make prostatic fluid (acinar adenocarcinoma), as well as the tubes of the prostate gland (ductal adenocarcinoma).
- The development of cancerous tumours is a multi-stage process, whereby successive genetic events occur in a normal cell to render it malignant.
- Early stage - The earliest prostatic lesion is prostatic intraepithelial neoplasia (‘PIN’), which is defined by neoplastic (abnormal) growth of epithelial cells within benign prostatic acini or prostatic ducts. PIN is not itself prostate cancer but, due to the common presence adjacent to prostatic adenocarcinomas, PIN is presumed to be a premalignant lesion.
- Localised cancer - Localised prostate cancer is cancer that is located completely inside the prostate gland. It has not spread outside of the prostate gland or to any other parts of the body. This is also known as clinically localised prostate cancer (‘CaP’).
- Locally advanced cancer - In locally advanced prostate cancer, cancer has extended outside the prostate gland. The cancer may have spread into the tissue surrounding the prostate, the seminal vesicles, or lymph nodes within the pelvis (i.e. N+: see [52.ii)] below, cf. N0).
- Metastatic cancer - Metastatic prostate cancer means the cancer has spread from the prostate to parts of the body outside of the prostate and beyond the lymph nodes in the pelvic region. It most commonly spreads to bone, lymph nodes beyond those in the pelvis, and visceral structures (most commonly the lungs and liver). When prostate cancer is metastatic, it can generally no longer be cured but treatment, primarily with androgen deprivation therapy (‘ADT’), may help to relieve symptoms such as pain and/or prolong the patient’s life by preventing disease progression.
- The patient pathway consists of diagnosis and histologic grading (Gleason score) and TNM (tumour node metastasis) staging. This is a way of describing the size of the cancer and its extent and spread, and healthcare professionals use this to determine the type of treatment to offer to patients.
- Gleason scoring of prostate cancer is the gold standard for histologic grading. The Gleason system assesses both the predominant and secondary patterns of gland formation within a prostate sample. The Gleason system recognises that cancerous cells fall into 5 distinct patterns as they change from normal cells to cancerous cells. The cells are graded on a scale of 1 to 5. The pathologist looking at the biopsy sample will assign one Gleason grade to the most predominant pattern in the biopsy and a second Gleason grade to the second most predominant pattern. The two grades are then added together to determine the overall Gleason sum score.
- Gleason sum scores generally range from 6 to 10, with 6 being the lowest grade prostate cancer and 10 the highest grade. Gleason sum scores of 3+3 or 3+4 would denote mostly low grade / low risk cancer, whereas a score of 4+3 denotes intermediate risk cancer, and scores of 4+4, 4+5 and 5+5 all denote high risk prostate cancer.
- Patients with a Gleason sum score of 6 or less typically respond well to therapy, whereas patients with a score of 8 or more have a greater chance of metastatic progression and resistance to treatment in the longer term.
- The TNM staging system is the common language in which healthcare professionals who treat and investigate prostate cancer can communicate the cancer extent for individual patients as a basis for decision making on treatment management and individual prognosis. The TNM staging system stands for Tumour, Node, Metastasis:
i) T – refers to the extent of the tumour locally within the prostate and how far it has spread into nearby tissue - it can be 1, 2, 3 or 4. In general terms, a lower number indicates a smaller tumour PSA screening.
ii) N - refers to whether the cancer has spread to the lymph nodes within the pelvis - it can be between 0 (no lymph nodes containing cancer cells) and 3 (many lymph nodes containing cancer cells). In practice N is sometimes classified simply as N0 or N+ rather than using the numerical range, with N+ denoting disease that has spread to the lymph nodes within the pelvis, as it is determinative for the choice of treatment.
iii) M - refers to whether the cancer has spread to another part of the body - it can either be 0 (no distant metastases) or 1 (distant metastases, which is also termed M+). Cancer that has spread to lymph nodes outside of the pelvis is considered metastatic. Additionally, for cases where metastases are established, they are classified into subtypes of where metastases are found: M1a (non-regional lymph node(s), M1b (bone(s)), and M1c (other sites)).
- Localised prostate cancer itself does not usually cause symptoms until the cancer has grown large enough to put noticeable pressure on the urethra, which carries urine from the bladder out of the penis. Symptoms of prostate cancer can include needing to urinate more frequently, hesitancy in starting to urinate, straining to urinate, a feeling that the bladder has not emptied fully, blood in the urine or semen, or unexplained loss of erectile function.
- While metastatic prostate cancer can also present with urinary symptoms as described above, patients may also present with symptoms reflective of cancer deposits at various sites in the body such as back or bone pain. Metastatic disease may also be associated with non-specific symptoms such as tiredness, anaemia, loss of appetite or weight loss. Death from prostate cancer itself usually only occurs if the cancer has progressed to the metastatic phase.
- Prostate cancer only affects men and a limited number of people with disorders of sex development.
- In 2006, with the exception of skin cancers (of which most were relatively indolent basal cell carcinomas), prostate cancer was the most frequently diagnosed cancer in men in the United Kingdom, with an estimated 30,024 newly diagnosed cases in 2006. In 2006, prostate cancer was the second leading cause of cancer deaths in men, exceeded only by lung cancer. In England and Wales, 9,017 deaths were registered in 2005 with the underlying cause of death identified as prostate cancer.
- Prostate cancer incidence increases with age. Although only about 1 in 456 men under the age of 50 will be diagnosed with prostate cancer, the incidence dramatically increases to 1 in 54 for the age range 50 to 59, 1 in 19 for the age range 60 to 69, and 1 in 11 for men aged 70 and older. Nearly 60% of all prostate cancers are diagnosed in men over the age of 65.
- Complaints of symptoms indicative of prostate cancer are a frequent presentation by patients to general practitioners in primary care. If a patient is suspected to have prostate cancer, a general practitioner would routinely order a PSA test, conduct a physical examination and, if prostate cancer is suspected, refer the patient to a secondary care specialist. MRI (magnetic resonance imaging) of the prostate was available, but not routinely offered.
- In secondary care, urologists, being specialists in the urinary tract and reproductive organs, diagnose, stage, and treat patients with prostate cancer. The treatment options available to urologists comprise surgical and systemic therapies, the latter involving ADTs. Most initial ADT therapy of patients with prostate cancer would be administered by urologists. Urologists (particularly those with a sub-speciality interest in genitourinary cancer, i.e. uro-oncologists) generally have a keen interest in keeping abreast of research developments and emerging therapies. They would often take part in and take a leading role in the development and running of clinical trials in the prostate cancer field.
- There were several different types of treatments available for prostate cancer. The choice of treatment was mainly directed by the stage of the cancer. The commonly used treatments can be summarised as follows:
i) Active surveillance / watchful waiting, with regular testing of PSA.
ii) Radical prostatectomy - The main surgery for the treatment of prostate cancer is radical prostatectomy: the removal of the entire prostate gland and surrounding tissue, including the seminal vesicles. This surgery was performed as open surgery (radical retropubic prostatectomy). In the United Kingdom, laparoscopic (key-hole surgery) prostatectomy (without robotic assistance) was only offered in a limited number of hospitals through private funding. In 2006, Guy’s Hospital in London started offering privately funded laparoscopic robotic prostatectomy, the first hospital to do so in the United Kingdom.
iii) Radiotherapy delivered either by external beam radiotherapy, or as brachytherapy. External beam radiotherapy, usually used in combination with ADT (with ADT as a neo-adjuvant with or without subsequent adjuvant therapy), is suitable for patients who present with localised or locally advanced cancer. Brachytherapy is a type of internal radiation therapy, in which seeds, ribbons or capsules containing a radiation source are placed in or near the tumour. There are two techniques for placing brachytherapy:
a) Permanent seed brachytherapy, also known as low dose-rate (“LDR”) brachytherapy, where small radioactive seeds are inserted into the prostate where they remain and release a steady dose of radiation over several months. LDR brachytherapy is used in patients who present with low-risk or intermediate risk cancer (i.e. Gleason score of 6 or 7). LDR brachytherapy can be administered in combination with external beam radiotherapy, but not with ADT.
b) High dose-rate (“HDR”) brachytherapy involves inserting thin tubes into the prostate. A source of radiation is then passed down the tubes into the prostate for a few minutes to destroy the cancer cells, after which the source of radiation is removed. HDR brachytherapy is suitable for the treatment of high grade / high risk prostate cancer: it is used in combination with external beam radiotherapy, and also almost always used with ADT. HDR brachytherapy was a relatively uncommon treatment choice in the United Kingdom, compared to external beam radiotherapy and LDR brachytherapy.
iv) Chemotherapy - which was beginning to be used to treat metastatic HRPC.
v) Palliative Treatments to slow or prevent bone damage and fractures in patients with metastatic prostate cancer.
- For treatment purposes, prostate cancer can be broadly categorised as clinically localised or metastatic, and as hormone-sensitive (i.e. androgen dependent) or hormone-resistant (HRPC, i.e. cancers that had progressed despite initial androgen deprivation, also known as ‘CRPC’).
- Systemic treatment for prostate cancer, where appropriate, has long been some form of ADT. Androgen deprivation is intended to reduce the quantity of androgens available to bind to the AR, or antagonise the action of androgens in the body, slowing the spread of the disease.
- Early on in the development of treatments for prostate cancer (around the early 1900s), surgical removal of the testes or testosterone-producing tissue in the testes (surgical castration) or oestrogen therapy (from the 1940s) were the primary means of ADT.
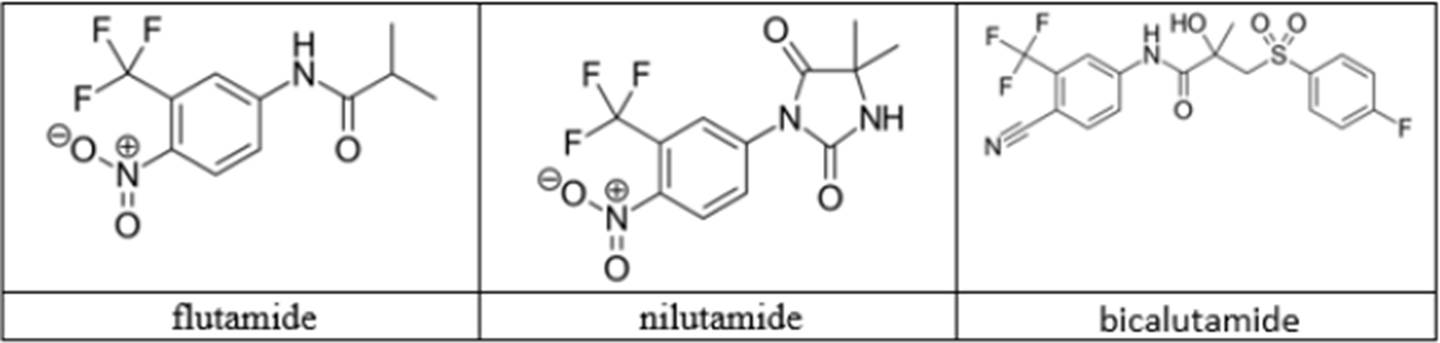
- Another approach to androgen deprivation is to use molecules that compete with androgens for binding with the AR and thus inhibit activation of the AR. Instead of agonising the AR, the molecules would prevent binding of the circulating androgen to the AR, thereby preventing its activation. This is referred to as AR antagonism. These types of molecules are known as antiandrogens or AR antagonists and the first treatment in this class was the steroidal antiandrogen cyproterone acetate or “CPA”, which came to market in the late 1970s, with later research leading to the development and approval of the non-steroidal antiandrogens flutamide, bicalutamide and nilutamide. Relevant details of these antiandrogens are set out in this table:
|
International
Non-proprietary
Name (brand name)
|
Date of first
approval
|
Steroidal?
|
Administration
|
Possible side effects
|
|
Cyproterone acetate (“CPA”) (Androcur/ Cyprostat/ Diane) |
Europe:
1978 |
Yes |
Tablets taken daily |
Loss of libido, erectile dysfunction, reduced sperm and ejaculate, hepatotoxicity, mood disorders, fatigue, dyspnoea cardiovascular adverse effects, gynecomastia and breast pain, hot flushes and sweats. |
|
Flutamide (Eulexin) |
USA: 1989 UK: 1997 |
No |
Tablet taken 3 times a day |
Gynecomastia and breast pain, erectile dysfunction, hepatotoxicity, hepatitis, nausea and diarrhoea, hot flushes, loss of libido, jaundice. |
|
Bicalutamide (Casodex) |
USA: 4 October
1995
UK: 18 June
2000 |
No |
Tablet taken
daily |
Loss of libido, erectile dysfunction, gynecomastia and breast pain, skin rash or itchy skin, hot flushes, nausea, jaundice, anaemia, mood disorders. |
|
Nilutamide (Nilandron/ Anandron) |
USA: 19 September
1996
Not widely used in UK |
No |
Tablet taken
daily |
Gynecomastia and breast pain, delayed adaptation to darkness, alcohol intolerance, diarrhoea, nausea. |
- Various combinations of treatments were available depending on the stage of the cancer and the risk category. Many of these involve ADT.
DEVELOPMENT OF ANDROGEN INDEPENDENCE
- As mentioned above, ADT lowers the level of circulating testosterone in the body, or in the case of antiandrogens, blocks the stimulatory action of testosterone. ADT had been shown to shrink prostate cancer tumours and reduce pain. In some patients, hormone therapy may slow disease progression for more than a decade; however, in others, it may keep the prostate cancer in check only for a few months. Eventually, prostate cancer cells can begin to resist the treatment.
- Prostate cancer has varying levels of androgen sensitivity as it progresses. Initially, tumours are similar to normal prostate epithelium and are androgen-dependent, so they usually regress in the absence of androgen.
- However, remaining tumour cells may either mutate, acquire a benefit or growth advantage, or have special characteristics that allow them to survive in a low-androgen environment. AR sensitivity or an AR mutation may allow for continued cell growth, leading to resistance to therapy developing, androgen-independent cells proliferating, and as a result to tumour growth and metastatic spread. Progression of metastatic disease begins to occur at a median of 13 months from the initiation of treatment with ADT.
- Cancers that progress despite initial surgical or medical castration are considered hormone-refractory (also referred to as castrate-resistant). Some patients with such tumours may respond to subsequent, different hormone therapy but this stage of the disease indicates cancer progression despite castration levels of testosterone.
- HRPC and CRPC are terms used interchangeably to describe these cancers that had progressed despite initial androgen deprivation. Other terms used in earlier literature and by some clinicians were “androgen independent prostate cancer” or ‘AIPC’. HRPC is the term commonly used by healthcare professionals.
NEW PROSTATE CANCER TREATMENTS AND EMERGING THERAPIES
- It was recognised by clinicians that, once prostate cancer developed to the metastatic stage, once ADT was instigated, the treatment effectiveness was time limited. New drug treatments are desirable in any disease. In the context of prostate cancer treatments (in particular, for the more advanced stages of the cancer, including HRPC) it was generally desirable to have treatments that were more efficacious than the existing standard of care, with more tolerable side effects, that provide improved overall survival, disease control, reduction in risk of progression and/or duration of response, and/or more optimal and convenient dosage forms, etc.
- New and more advanced treatment options for prostate cancer were also desirable and sought - which is the case for most if not every disease. There were several avenues of drug discovery and trials involving patients ongoing in the treatment of prostate cancer, many of which had shown promising results in pre-clinical studies such that clinical studies were initiated. There was a variety of research in the preclinical stage (i.e. in vitro and in vivo (animal studies)). Clinical research at the time included many hormonal and non-hormonal approaches (including immunotherapy (Onyvax-P and GVAX), chemotherapy (satraplatin, paclitaxel and docetaxel), and other therapies that acted on specific biological targets (such as the 5α-reductase inhibitors finasteride (Proscar, Propevia) and dutasteride, which were already in use for benign prostate hyperplasia)). Clinicians would have been aware of certain developments in clinical research in their day-to-day practice.
- PSA is a protein that is made predominantly in the luminal epithelial cells of the prostate gland and is a well-known prostate tumour marker used in clinical practice for prostate cancer detection and the monitoring of treatment. It is found mostly in semen, with very small amounts released into the bloodstream. When there is a problem with the prostate (e.g. infection, inflammation or enlargement (by benign hypertrophy)), more PSA is released. Higher than average age-specific mean PSA levels are a good indicator of prostate cancer, and in the later stages of prostate cancer, PSA can be raised to very high levels. PSA can be measured in a blood sample from the arm. PSA screening can help identify prostate cancer at an early stage when treatment may be more effective and potentially have fewer side effects. PSA tests are routinely ordered in primary care for patients presenting with symptoms associated with prostate cancer. Additionally, in secondary care, PSA is used as a diagnostic tool with symptomatic patients, and as a tumour marker, measuring the response to treatment of patients with a diagnosis of prostate cancer.
- Changes in PSA levels can be used to track disease regression and progression. An increase in the serum PSA level is associated with progression of prostate cancer and a decrease in PSA with disease regression. A drug that decreases secreted PSA levels could potentially be useful as a treatment for prostate cancer.
- Increased androgen or AR can result in too great a growth of prostate cells and ultimately can lead to loss of growth control and cancer. This link, between androgen and prostate cancer, has been known for decades and resulted in the development of ADT, as summarised above, that target this growth signalling pathway.
- Anti-androgenic approaches to treatment included administration of suitable drugs that compete with androgens for the AR ligand-binding domain (“LBD”) and prevent activation of the AR. This “competitive binding” would therefore block the AR in the presence of normal circulating androgen levels. Of these, bicalutamide (the classic AR antagonist) was considered the gold standard treatment for prostate cancer. In basic studies of androgen response to treatment in laboratory settings it could be used as a comparator drug against new hormonal therapies and it was in common use in clinical practice and a variety of prostate cancer trials.
- The available anti-androgenic therapies for treatment of HSPC were widely used and clinically useful agents, but they did not prevent prostate cancer progressing from the hormone sensitive to the hormone refractory state. Moreover, they were ineffective in treating HRPC as the drugs lost their anti-androgenic activity and could become agonists, stimulating further progression of the disease rather than inducing its regression. There was no definitive evidence for one predominant cause to explain the progression from HSPC to HRPC. Some of the theories for progression were mechanisms that were not expected to be resolved by anti-androgens. Research at the time therefore included a variety of hormonal and non-hormonal approaches.
- Among the prominent theories as to the mechanisms that might be involved in progression of HSPC to HRPC were:
i) The development of mutations in the AR gene allowing for activation by non-androgen ligands (including antiandrogens such as bicalutamide);
ii) Ligand-independent activation of AR through ‘cross-talk’ with other signalling pathways;
iii) Altered expression of coregulatory molecules affecting regulation of DNA binding;
iv) Increased AR gene expression leading to increased sensitivity of prostate cancer cells to low levels of androgen; and
v) Incomplete depletion of androgen in the tumour tissue despite castrate levels in the blood.
- It was postulated that some of these mechanisms could provide a survival and growth advantage in an androgen depleted environment, and thus become established in prostate cancer tumours via clonal selection.
- Considering the understanding of the potential mechanism(s) for progression to HRPC, the therapies being studied at the time spanned a range of targets. AR was recognised as a central node of this cancer type and was a focus for development of new cancer therapies. There were a number of avenues being investigated with a view to targeting that axis to treat hormone refractory disease. Among the avenues under investigation were:
i) Approaches that targeted the expression of AR itself, for example RNA interference or antisense oligonucleotide approaches;
ii) Approaches that targeted the interaction of AR with the cofactors implicated in activation or suppression of transcriptional complexes;
iii) Approaches that targeted the AR LBD, including attempts to identify new antiandrogens with reduced agonist effects;
iv) Approaches that targeted other signalling pathways to prevent ligand-independent activation of AR; and
v) Approaches that sought more effectively to reduce the levels of testosterone and DHT in the bloodstream and prostate tissue.
- There was no single cell line or animal model that reflected all the aspects of prostate cancer in humans. Cell-based studies are generally the first type of assay to be conducted on new drug candidates and positive results are used to determine if a drug candidate is promising enough to be subjected to in vivo studies. Mouse models of prostate cancer, such as those using xenografts of human prostate cancer cells in SCID (severe combined immunodeficiency disease) mice were known and widely used.
- A number of in vitro and in vivo assays were used to assess a compound for its potential effect on prostate cancer. With each assay, it is common to compare the test compound against a drug with a known behaviour, for example the anti-androgen bicalutamide. The synthetic AR agonist, metribolone (R1881), which is a ligand and agonist of the AR, is also used in research as a testosterone analogue to stimulate prostate cancer cellular growth.
- There are many cell lines used in prostate cancer research. The most commonly used cell lines at the Filing Date were LNCaP, DU145, PC-3 or variants thereof. LAPC4 is a further cell line of human prostate cancer which was developed from tissue taken from men with HSPC who were undergoing surgical procedures.
- The cell line LNCaP (Lymph Node Carcinoma of the Prostate) is an epithelial cell line derived from a metastatic lymph node of a patient with prostate adenocarcinoma.
- LNCaP cells express an endogenous level of functional AR and are androgen sensitive (so they can be stimulated by the presence of androgen or alternatively their function can be inhibited by an anti-androgenic agent). LNCaP cells also express PSA. Owing to those properties the cell line was used commonly in laboratory testing as a model of HSPC. The effect of a putative antagonist or agonist in this hormone sensitive model could be assessed, for example, by measuring the PSA level produced by the cell line with the test compound present versus the PSA level produced by the cell line against a suitable control (for example, without the test compound being present or in the presence of an anti-androgen such as bicalutamide).
- The LNCaP cell line could be modified to make the cells overexpress the AR. This modification could be achieved by growing LNCaP cells in a low androgen environment. Over time, the cells would respond to the low androgen environment.
- The environment of LNCaP cells may also be altered to test the antagonist or agonist properties of a compound. The cells could be grown in androgen replete media (such as foetal bovine serum, “FBS”, which provides normal levels of androgen), or in androgen deplete media (such as charcoal stripped bovine serum, “CSS”, where androgen is stripped out to represent castrate levels of androgen). LNCaP cells cultured in androgen replete media are used to test for antagonism: these cells are hormone sensitive (as explained above) and are induced to proliferate by the presence of excess androgen in the culture medium. Thus, if a test compound reduces or abolishes cell growth, the test compound is understood to be antagonising the AR. Conversely, in androgen-deplete culture medium, the LNCaP cells are not stimulated by androgen. Accordingly, they would not be expected to proliferate. If the test compound causes an increase in cell growth, the compound is understood to be stimulating (agonising) the AR.
- PSA measurements. PSA concentration is commonly tested using enzyme-linked immunosorbent assays (“ELISA”), which are available in standardised kits. ELISA is an immunoassay which detects and amplifies antigen-antibody reactions by using covalently bound enzyme-antibody molecules. In this type of assay, the antigen (the target macromolecule) is immobilized on a solid surface (often a microplate, sometimes via a capture antibody fixed to the microplate: a sandwich ELISA) and then complexed with an antibody that is linked to a reporter enzyme (sometimes, but not always, via a secondary antibody). Unbound antibody must be washed away adding to the number of steps. The choice of antibody used determines the specificity of the ELISA assay. In the field of prostate cancer, it was common to detect PSA protein, which would require an antibody specific to PSA. This assay could confirm protein had been translated from the mRNA (other methods, such as micro-arrays, measure expression at the RNA level). Detection is accomplished by measuring the activity of the reporter enzyme via incubation with the appropriate substrate to produce a measurable product (the enzyme converts the substrate to a product that can be detected, usually with quantitative colorimetric methods). The bound protein can be referenced to a standard curve of known concentration and the amount of PSA produced quantified. The ELISA may be used on the output of a cell-based assay but can also be used on a range of other sample types. They are simple and routine assays used widely in laboratories.
- Luciferase reporter assays are used to assess transcriptional activity in a cell transfected with a reporter construct containing the response element of a gene of interest. For example, cell lines expressing AR (either naturally such as LNCaP, or where recombinant AR is introduced) may be engineered to incorporate an androgen response element driving downstream reporter gene expression (e.g. luciferase). In practical terms, the upstream promoter of an AR responsive gene (to which AR binds, e.g. from the PSA gene) is cloned ahead of the gene for luciferase and introduced to a cell line to create a modified cell line. The luciferase reporter is expressed when the target protein (AR) is activated. The luciferase protein produces light by enzymatic action on a substrate, proportional to the level of its expression. The assay may thus be used to detect the level of AR activation. Luciferase assays are usually reported as relative values.
- MTS assays. These measure mitochondrial activity, which can be used as a surrogate of cell growth, proliferation and cytotoxicity. MTS assays are available in commercial kits, whose test results are generally displayed as “relative units”. MTS assays may be used, for example, to indirectly measure the growth inhibition of cells when subjected to an inhibitory compound.
- Competition binding assays measure the ability of labelled ligand to bind specifically to a target protein in the presence of a second competing but unlabelled ligand. In prostate cancer research this assay is used to assess the binding affinity of a compound to a receptor/protein of interest in comparison to another compound.
- Western blotting. A western blot visualises and approximately quantifies proteins of interest. Western blots are often used in research to separate and identify proteins. In this technique a mixture of proteins is first separated, usually by gel electrophoresis based on molecular weight. The proteins in the gel are then transferred to a membrane (the “blot”). The membrane is then incubated with antibodies specific to the protein(s) of interest. The unbound antibody is washed off leaving only the antibody bound to the protein of interest. The bound antibodies are then detected (the method can use a range of different labelling approaches). As the antibodies only bind to the protein of interest, usually only one band should be visible. The thickness of the band corresponds to the amount of protein present; thus, comparing to an internal standard (β-actin is commonly used) can indicate the relative amount of protein present.
- Where the drug candidate demonstrates cellular activity, the Skilled Cancer Biologist would progress the compound to in vivo testing. Animal models are used in later pre-clinical drug development to investigate how a drug will act in a living system. Prior to clinical testing, a drug candidate would be tested in in vivo assays.
- In vivo treatment model assays included subcutaneous xenografts. Subcutaneous xenograft models are used to study tumour growth and response to chemical compounds. These types of in vivo models either involve injecting tumour cells into the flank of the mouse, waiting for the tumour to develop and then begin treatment or involve implanting tumour cells into the flank of the mouse and immediately delivering treatment.
- At the outset of a drug discovery project, the Skilled Team would typically have in mind an intended approach to treating the disease of interest. This would typically be led by the Skilled Cancer Biologist, who would lead with the concept of how the disease would be targeted - that is, what biological pathway would be sought to be targeted, and which proteins or receptors or other biological features need to be modulated in order to achieve that biological effect.
- With the intended biological target in mind, the Skilled Team would identify a target product profile (“TPP”) for their prospective therapeutic compound. This is a description of the properties of the compound which the Skilled Team would be seeking to identify. The properties identified in this profile would typically include physicochemical properties (e.g. appropriate molecular weight and lipophilicity), biological properties (e.g. activity against the target and minimal off-target effects), pharmacokinetic and pharmacodynamic properties (e.g. appropriate half-life), and drug-like qualities (e.g. not toxic at therapeutic doses).
- The TPP would typically be framed in a discussion between the Skilled Medicinal Chemist and Skilled Cancer Biologist. The Skilled Cancer Biologist would typically be responsible for identifying the biological target and the conceptual approach to therapy. The Skilled Team would then discuss the properties of a compound which would be suitable for therapy, and so draw up their TPP. Once that profile had been determined, it would enable the Skilled Medicinal Chemist to commence work to synthesise candidate compounds, propose changes to compounds of interest, and test their properties against the TPP. The Skilled Team may revisit the TPP if, following initial attempts, it appears unobtainable or turns out to be unsuited for the intended therapy.
- With the TPP in mind, the Skilled Medicinal Chemist would seek to identify a compound or collection of compounds, which are a starting point for improvement. There are broadly three different starting points in a drug discovery programme: starting from compounds published in the literature, starting from the endogenous ligands (i.e. looking at what ligand binds to the target in the body), or by high throughput screening.
- In a field with a number of compounds published in the literature, the Skilled Medicinal Chemist would wish to understand whether the compounds represent credible starting points for a new drug discovery initiative. A common first step would be to analyse the reported data concerning those compounds, typically by reviewing relevant biological data (e.g. potency, selectivity, or pharmacokinetics data as may have been published). The Skilled Medicinal Chemist would also consider whether the compound(s) look like molecules that have the potential to be optimised to a potential drug molecule.
- To find a starting point in literature, the Skilled Medicinal Chemist would use a tool like SciFinder or CrossFire (which was later re-branded to Reaxys) to locate relevant journal papers, conference abstracts, and patent literature. The Skilled Medicinal Chemist might also search using other sources like PubMed.
- If a suitable starting point is identified, the Skilled Medicinal Chemist would most likely adopt an iterative approach to modify the starting point molecular structure to try to improve its activity, selectivity and/or physicochemical properties, having in mind the TPP. Iterative modification of a compound is done by developing a molecule that has a structure partially similar to the starting compound but with some different chemical substituents or modifications (i.e. a structural analogue). In order to progress the drug design process, the Skilled Medicinal Chemist often builds up a structure-activity relationship (“SAR”) library of such compounds and their test data. The Skilled Medicinal Chemist could also supplement their SAR work using computer modelling and, if available, protein-ligand structures (such as by X-ray crystallography).
- To do this, and as a very broad description of the general steps involved in the process of drug discovery, the Skilled Medicinal Chemist would ordinarily make a series of chemical modifications to the starting compound, resulting in a number of different structural analogues. The Skilled Medicinal Chemist is trained in methods of adding functional groups to compounds, converting functional groups, and carrying out coupling reactions. They would use commercially available catalogues for the molecular “building blocks” as well as materials to effect synthetic transformations such as solvents, acids and bases. These analogues would be tested to determine how each of the modifications affects their properties, including activity against the target, selectivity (by measuring activity against other off-targets), solubility, permeability, etc. Through this process, the Skilled Medicinal Chemist would build up an idea of which parts of the compounds and what types of substituent modifications impact the relevant properties (such as binding, efficacy, physicochemical properties, metabolism, etc.), and the size and nature of each of those modifications.
- The next broad step in the drug discovery process is to use in vitro and/or in vivo models that are representative of the disease. The biological testing of the molecules would be conducted by other members of the Skilled Team, although the Skilled Medicinal Chemist would have a general understanding of many of the tests and their parameters and outputs.
- Within a molecular drug candidate structure, it is usually possible to identify a molecular ‘scaffold’. This is the core chemical grouping of the molecule around which different substituents can be fixed. A Skilled Medicinal Chemist would be able to identify the scaffold of a molecule by looking at the structure. Drug candidate scaffolds are usually based on a single or multiple carbon-based ring system, which can be aliphatic or aromatic, and can optionally contain so-called heteroatoms (e.g. nitrogen, oxygen or sulphur) in place of selected carbon atoms around the ring.
- The position of substituent groups on a scaffold, for example alkyl substituents, may be referred to as vicinal or geminal. For example, two methyl groups on adjacent carbons of a scaffold would be referred to as vicinal whereas two methyl groups on the same carbon would be referred to as a geminal di-methyl substitution. An example is provided below based on a simple ring system (cyclohexane) to illustrate this point.
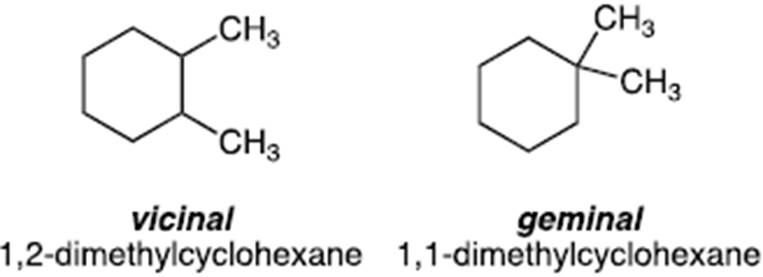
- The groups which can be added in a geminal arrangement may be limited by size.
- Furthermore, it is not possible to have geminal groups directly attached to an aromatic ring as it would violate the rule that carbon atoms must form four bonds. Therefore, it would disrupt the stability of the ring.
- An alkyl group is a chain of carbon and hydrogen atoms connected by single bonds - a ‘hydrocarbon’ chain.
- The nomenclature for these depends on the length of the carbon chain. Methyl (one carbon), ethyl (two carbons), propyl (three carbons), butyl (four carbons) etc.
- Alkyl chains may be linear or branched. Take, for example, a butyl group, which consists of four carbon atoms and nine hydrogen atoms (C4H9). These can be arranged with three carbon atoms bonded to one another via single bonds, each with two bound hydrogen atoms plus a terminal carbon atom with three bound hydrogen atoms (known as ‘normal’ or n-butyl). Alternatively, there may be a central carbon atom with the remaining three carbon atoms bonded directly to this. Each of those carbon atoms are bonded to three hydrogen atoms. This is known as a tertiary, or tert-butyl group. There is also an intermediate branched butyl structure known as a secondary, or sec-butyl group.
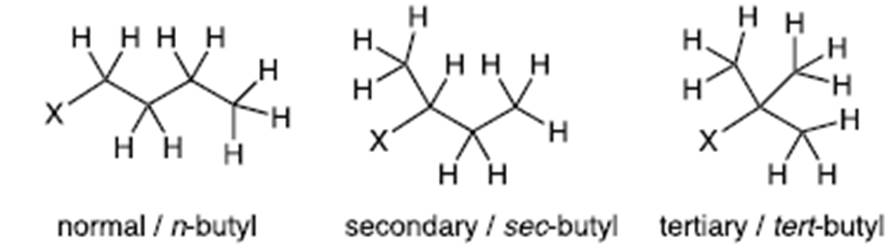
- Branching of alkyl chains affects the amount of space which the alkyl groups take up.
- There is no limit on the length of hydrocarbon chain.
- A carbon atom may bond to another carbon via a double bond or a triple bond. These structures are referred to as alkenes and alkynes, respectively.
- Alkenes and alkynes are referred to as ‘unsaturated’ carbon chains, whereas alkyls are known as ‘saturated’ carbon chains.
- Alkyl groups may also be joined together at either end to form a ring structure, known as a cyclic structure. Cyclic structures may be formed of saturated or unsaturated carbon chains, a fully unsaturated ring is called an aromatic (or aryl) ring (e.g. phenyl (Ph)).
- A cyclic structure may be present as a substitution to an alkyl chain or another ring structure. When substituted onto another ring the cyclic alkyl can be linked by a single bond or as a ‘spirocyclic’ group. In the latter, two cyclic hydrocarbon groups are bonded at a shared carbon ‘pivot’ point to form a twisted structure.
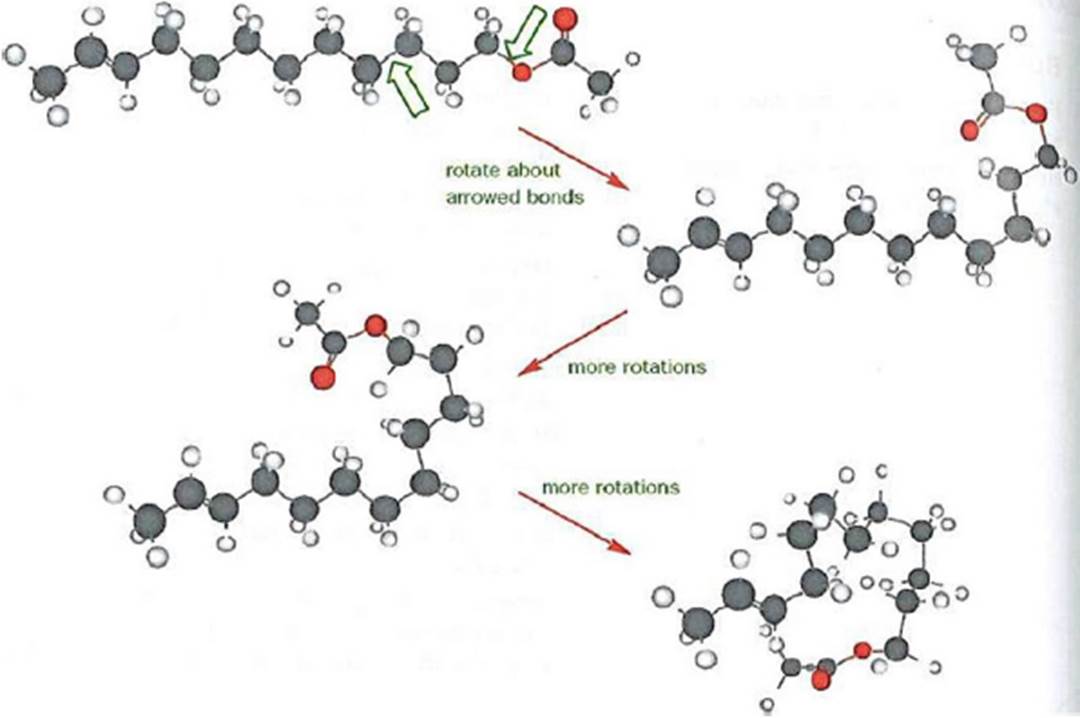
- Rotation is possible about single covalent bonds and this rotation means that the molecule as a whole can adopt a number of different shapes, even though the atom-to-atom connectivity remains the same. This is also related to the concept of a molecule’s rigidity. A benzene substituent (also known as a phenyl group) is an aromatic ring that is rigid. An alicyclic compound (e.g. a cyclohexane) may have some conformational flexibility and be less rigid than benzene. A non-cyclised aliphatic chain (e.g. a hexyl group) has conformational freedom, particularly if it is a long-chain aliphatic compound, which gives rise to the possibility of torsional flexibility. In other words, whilst the aliphatic chain may be represented as a relatively straight (zigzag) line in a 2-dimensional chemical drawing, in a real world setting all the single bonds in the molecule can constantly rotate, resulting in the molecule adopting different “bent” or “folded” shapes. This is illustrated in the below Figure. (Clayden, J. et al. (2001). Organic Chemistry. Oxford University Press.)
- A chiral centre is an asymmetric carbon (four different substituent groups bonded to the one carbon). Chiral comes from Greek for hand/ handedness. And where you have a chiral centre you have a mirror image, a non-superimposable image. Such a molecule is called an enantiomer. Chirality is essential for life; our constituent proteins and nucleic acids (DNA/RNA) are naturally chiral. Here, we focus on the issue of chirality in synthetic drug candidates, which may or may not have a chiral centre(s). Although chemical structures are often drawn on 2D surfaces (e.g. paper), it is important to represent structures in their true 3D form. Chemists therefore use a ‘wedge’ bond to denote atoms projecting out of the paper, and a ‘dashed’ bond to denote atoms projecting back into the paper. A simple chiral centre is denoted below, with the two enantiomeric forms shown.
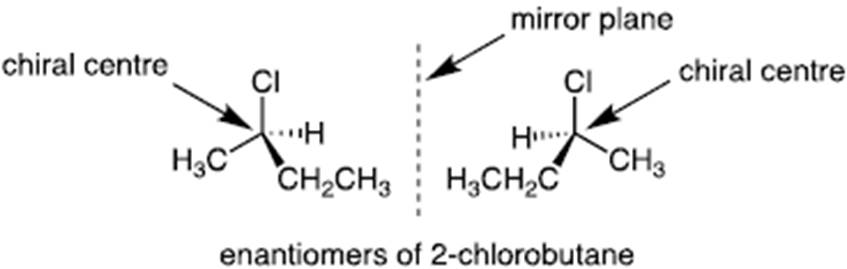
- It is possible to have one or more chiral centres in a molecule. Where there is just a single chiral centre, there would be two forms of the molecule and the forms would be mirror images of each other but non-superimposable. The two enantiomers would be predicted to have the same physical characteristics (e.g. melting points and hydrophobicity (explained in [131] below)) but they may have different biological activity. One may have a therapeutic activity and one may have a toxic effect. It would only be possible to know this by testing.
- When you synthesise a molecule with chirality one way is to create a racemic mixture (containing both enantiomers). However, it is possible either to synthesise a specific enantiomer (known as asymmetric synthesis) or to separate them out once synthesised so that the enantiomers can be tested. Asymmetric synthesis or chiral separation may be technically challenging, especially at large scale when dealing with later-stage drug candidates. A further alternative is to start the chemical synthesis with a commercially available enantiomerically pure building block, such as an α-amino acid.
- There are lots of drugs with chiral centres. Including a chiral centre within the design of the molecule may add to the complexity of the drug development process.
- The steric properties of the molecule concern its size and shape. Steric hindrance is a consequence of insufficient physical space in the binding pocket.
- The steric effects of a molecule can be determined empirically or through computational analysis. In pharmaceutical chemistry, the steric properties of a functional group within a molecule can be increased or decreased by changing the length of a carbon chain, or by increasing the size of a ring structure.
- It is important for the Skilled Medicinal Chemist to consider the stability of the compounds. This includes stability of the drug as it passes through the gastrointestinal tract but also metabolic stability in the liver.
- The Skilled Medicinal Chemist would know that adding a fluorine was one approach to try preventing metabolic oxidation of a phenyl ring.
- Understanding SAR is the key to designing new analogues with better properties. The medicinal chemistry approach to SAR is based on comparisons of structural changes with activity changes. By changing discrete parts of a chemical structure in a systematic way, the Skilled Medicinal Chemist tries to build up an idea of which parts of the molecule are important for which biological aspects because the central axiom of SAR is that the activity or property of any given molecule is related to its structure.
- Additionally, high resolution X-ray co-crystal structures and models of analogue series members bound to the target protein may be used in some cases to interpret SAR and help guide new, testable design hypotheses.
i) X-ray crystallography is a biophysical technique for determining protein-ligand interactions, which can be used to supplement SAR work. This technique provides high resolution atomic coordinates for directly visualising protein-ligand interactions. However, X-ray crystal structures for certain drug discovery targets may not always be available. Where available, consistent, iterative feedback of new co-crystal structures determined with the candidate drugs complexed with the target protein can be a valuable addition to a drug discovery programme.
ii) Candidate drugs can also be computationally docked into an X-ray crystal structure model to predict binding interactions and ligand fit. Such computational methods are still inadequate to predict potency with sufficient accuracy to drive medicinal chemistry in most cases, but they can provide useful qualitative guidance.
iii) For a new target protein with unknown 3-dimensional structure, its structure could be predicted from the crystal structure of a highly similar protein. Such modelling of the structures of new proteins by homology is called homology modelling. This can sometimes provide structures that are helpful, but (where available) co-crystal structures of relevant ligands bound to the target protein are much more valuable.
- There are a variety of interactions that occur within and between molecules and that can be formed between a ligand and the target.
- Intramolecular forces. Intramolecular forces are the chemical bonds that hold the atoms together in a molecule. They include (i) metallic bonds (electrostatic interactions between electrons and positively charged metal ions), (ii) ionic bonds (electrostatic interactions between ions with large electronegativity difference), and (iii) covalent bonds (bonds in which two atoms share pairs of electrons, e.g. a single bond involving two shared electrons).
- Intermolecular forces. Intermolecular forces (forces between two or more molecules) are weak relative to intramolecular forces. They arise in interactions between molecules (drug-drug or drug-target) and the number and type of these interactions is dependent on the structure of the drug and the functional groups that are present. Intermolecular forces include:
i) Electrostatic or ionic bonds. These are the strongest intermolecular bond and take place between groups having opposite charges.
ii) Van der Waals forces (which act between most molecules). This term is often used to describe distance-dependent forces between molecules that are not covalent or ionic chemical bonds.
iii) Dipole-dipole interactions (which act between polar molecules). Many molecules have a permanent dipole moment resulting from the different electronegativities of the atoms and functional groups present. The strength of the dipole-dipole interactions falls away more quickly with distance than electrostatic interactions but less quickly than Van der Waals interactions.
iv) Ion-dipole interactions (which act between ions and polar molecules).
v) Hydrogen bonding (which acts between an electronegative atom (usually N, O or F) and a H atom attached to another electronegative atom). They are usually regarded as strong non-ionic interactions and usually take place between an electron-rich heteroatom (usually oxygen or nitrogen; sometimes sulphur or fluorine) and an electron-deficient hydrogen.
vi) Repulsive interactions occur if molecules come too close, and repulsive steric and electronic (i.e. two positive or two negative charges) forces clash.
- Hydrophobic interactions are also another type of intermolecular force. These interactions arise when a hydrophobic (“water hating”) molecule is placed in an aqueous environment. There is an energetic penalty when the hydrophobic portions of the molecule are exposed to water molecules in an aqueous environment. That penalty can be reduced by the molecule folding in such a way that the hydrophobic regions come into close physical proximity with other hydrophobic regions of the same molecule or other molecules so that, together, they avoid being exposed to the polar solvent, water. Hydrophobic regions may comprise certain amino acids that have hydrocarbon side chains that are non-polar. Hydrophobic interactions are important for protein folding. The strength of hydrophobic interactions is related to the number of carbon atoms within a molecule (or within the interacting region). A greater number of carbons typically results in stronger interactions. It will also depend on the fit between the interfaces.
- The properties identified in the TPP are set out at paragraph 96 above. Relevant concepts are as follows.
- The physicochemical properties of the molecule may be computationally predicted and/or experimentally measured. The primary physicochemical properties of interest include:
i) Solubility and permeability. A drug substance often needs access to a patient’s circulation to be delivered to the site of biological action. A drug may be injected directly into the patient’s bloodstream or administered orally, in which case it has to be absorbed from the digestive system. This requires that the drug be soluble and, for an oral drug, that it permeates the gastrointestinal membranes.
ii) Partition coefficient (“P”) describes the equilibrium solubility of a drug in an immiscible biphasic system of water and a lipophilic solvent, often 1-octanol. P is defined as the ratio of the drug concentration in the lipophilic phase divided by the drug concentration in the aqueous (water) phase. The drug’s “lipophilicity”, or hydrophobic character, can be expressed as the logarithm of the partition coefficient (“log P”). Positive log P values indicate that the drug is more soluble in the lipophilic phase and the larger the log P value the more lipophilic the molecule is. A drug’s lipophilicity can influence a number of its properties, including solubility, permeability, etc.
iii) The degree of ionisation (“pKa”) is a measure of the acidity of a drug in solution and can be a useful parameter in understanding the behaviour of drug molecules. A lower pKa indicates that the drug is a stronger acid.
- Toxicities. The Skilled Medicinal Chemist would also test for toxicities, and there would be certain groups that the Skilled Medicinal Chemist would avoid due to potential toxicities.
- Binding affinity. The binding and dissociation of a ligand (or a drug) with a receptor can be described as an equilibrium between bound and unbound protein and ligand. In drug development, the binding affinity for a new ligand to a receptor is commonly measured in comparison to an established ligand that is known to bind to a receptor with high affinity in competitive binding studies.
- Half maximal inhibitory concentration (“IC50”) is a measure of potency, and is a quantitative measure that indicates how much of a particular inhibitory substance is needed to inhibit the activity of the target in a given biological process by 50%. IC50 values are typically expressed as molar concentrations. The IC50 of a drug can be determined by constructing a dose-response curve and examining the effect of different concentrations of an antagonist on inhibiting activity. IC50 values are used to compare the potency of two or more antagonists in development, or against an approved drug. A numerically lower IC50 value indicates a higher potency.
- ADME is a framework of concepts commonly used to help guide drug optimisation. The components of ADME are as follows:
i) Absorption refers to the drug entering the systemic circulation (bloodstream) from the site of administration. For orally administered drugs, absorption is a primary focus as a drug must be absorbed from the gastrointestinal tract before it can have a medicinal effect in the body. Changes in absorption have significant effects on a drug’s PK profile.
ii) Distribution refers to how a drug spreads throughout the body. The distribution is dependent on factors, including vascular permeability, blood flow, ability of the drug to bind to tissue and plasma proteins, and its lipid solubility.
iii) Metabolism refers to biotransformation of a drug by organs or tissues so that the drug can be excreted, including the rates and pathways of those biotransformations. To facilitate removal via faeces or urine, the drug compound may be altered to be more water-soluble. A commonly encountered problem early in lead optimisation is the need to make a molecule more metabolically stable.
iv) Excretion consists of pathways that remove an administered drug and/or its metabolites from the body.
- Pharmacokinetics (“PK”) refers to how the body affects a specific substance after administration. PK is important in the development of drugs as part of understanding whether they will be safe at the appropriate dose and maintain efficacy for the desired amount of time. Through in vivo experiments and mathematical modelling, ADME characteristics discussed in the preceding paragraph are investigated to understand a drug’s PK. One technique is to measure the relationship between drug plasma concentration and time elapsed since drug administration. From that relationship, numerous PK parameters can be determined, including the following:
i) Cmax is the maximum (peak) concentration that a drug achieves in the blood after the drug has been administered.
ii) Tmax is the period of time it takes for a drug to reach the peak blood plasma concentration following administration. Tmax is the time at which Cmax is observed.
iii) Area under the curve (“AUC”) is an estimation of the total amount of the drug absorbed into systemic circulation after administration within a defined time period.
iv) Half-life refers to the amount of time required for a drug’s concentration in plasma (or other material) to decrease by 50%.
v) Clearance refers to the volume of biological media cleared of drug per unit time.
vi) A steady-state concentration (“Css”) is achieved when the amount of a drug being absorbed is the same amount that is being cleared from the body when the drug is given continuously or repeatedly.
- Pharmacodynamics (“PD”) refers to what a drug does to the body and considers the biochemical, physiologic and molecular effects of drugs and metabolites, and the relationship of these processes to pharmacological effects (both therapeutic and toxic). PD assays would be performed by other members of the Skilled Team and the Skilled Medicinal Chemist would review the data from such assays and use it to inform their work.
- The Skilled Team would have known that the AR was a member of the nuclear receptor superfamily, which shared basic structural and functional homology. They would also have known that members of this superfamily were ligand dependent nuclear transcription factors and consisted of three basic functional domains: the DNA binding domain, the ligand binding domain and the amino terminal domain.
- The mechanism of action of AR antagonists (preventing the interaction of testosterone and DHT with the AR) would have been known to the Skilled Medicinal Chemist, as was the fact that there were two broad classes of such antagonists: steroidal derivatives and non-steroidal derivatives.
- The structures of certain AR antagonists would have been known to the Skilled Medicinal Chemist or could easily have been looked up if required. Of the non-steroidal AR antagonists, they would include flutamide (marketed as Eulexin), nilutamide (marketed as Anandron) and bicalutamide (marketed as Casodex).
- Once pre-clinical drug development is complete and a suitable lead candidate has been identified by in vitro and in vivo experimentation, it progresses through phase I, II and III clinical trials. Phase I trials are “first in man” studies and are intended to determine an acceptable and non-toxic dose in humans, assess the metabolism and safety of the compound, and if possible, to ascertain whether the drug under test is active in the human body. Pre-clinical studies therefore set the scene for these parameters being met. Phase II studies are generally used to determine the activity of the drug in the intended patient group and to further assess toxicity. Phase III studies are large, randomised studies that are used to compare relative efficacy and toxicity of the new drug candidate when tested against the available treatments, at larger scale.
- At the end of the Statement of Agreed CGK, the parties identified one point in dispute: the CGK regarding the metabolic stability of terminal dimethyl groups. This dispute is best considered in the context of the arguments on obviousness.
- The closing submissions also revealed some additional mini-disputes regarding CGK points. Again, these are best resolved in the obviousness sections.
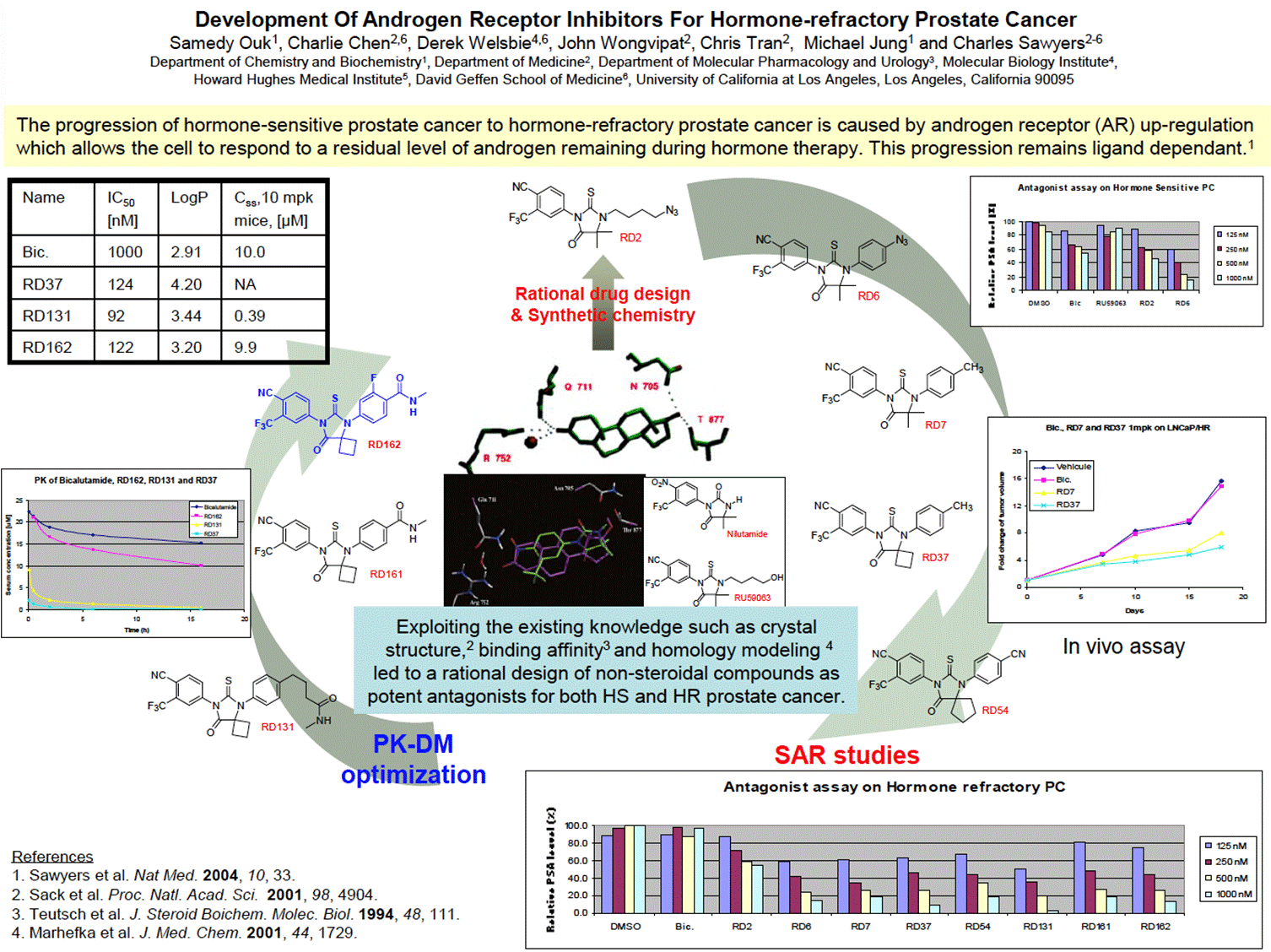
- The Poster is headed “Development of Androgen Receptor Inhibitors for Hormone-Refractory Prostate Cancer”. It describes what it calls a “rational design” of “non-steroidal compounds as potent antagonists for both HS and HR prostate cancer”. It is well designed and conveys a large amount of useful information to the Skilled Team. By way of overview, the Poster looks like this:
- Professor Ward said that the Skilled Medicinal Chemist would understand that “rational design” involves making a series of structural changes to a compound and studying the resulting changes to its properties to try to develop an understanding of the SAR.
- The Poster depicts the step-wise process of development undertaken by the inventors, starting with RD2 at the top and flowing clockwise around the SAR studies undertaken to the most potent compound following the SAR (RD37) through the PK optimisation to arrive at RD162. A skilled chemist can see from the numbers used for the compounds that not all are depicted and would understand that many more are likely to have been made and tested.
- Under the heading is a yellow box with a two-line summary explaining that AR upregulation is responsible for the progression of hormone-sensitive to hormone-refractory prostate cancer. This was one of the known explanations for why this progression occurred (see [78.iv)] above. In footnote 1 there is a cross reference to a prior paper by the Sawyers group published in Nature Medicine 2004 (this paper is attributed “Sawyers” as last-named author, but in the Slides and elsewhere to “Chen” as first-named author, but it is the same paper).
- In the central blue box, it is explained that “Exploiting the existing knowledge such as crystal structure,2 binding affinity3 and homology modeling 4 led to a rational design of non-steroidal compounds as potent antagonists for both HS and HR prostate cancer”.
- “HR” prostate cancer would be understood to be shorthand for hormone-refractory prostate cancer and “HS” prostate cancer to be referring to hormone-sensitive prostate cancer.
- The structures of nilutamide (a well-known antiandrogen used at the Application Date in the treatment of prostate cancer) and a compound called RU59063 are shown in the white central box.
- Footnote 2 is the Sack paper, footnote 3 is the Teutsch paper, and footnote 4 is the Marhefka paper. It is common ground that the skilled reader would have regard to the cross-referenced papers. On doing so, they would see that:
i) the crystal structure shown in the black box in the centre of the Poster is a reproduction of Fig 10 of Marhefka. This shows the binding model for nilutamide (in green) superimposed on testosterone (in purple) within the human AR ligand binding domain.
ii) RU59063 is described in the Teutsch paper as having high binding affinity to AR and in vivo antiandrogenic activity.
iii) The figure in the centre of the Poster is a reproduction of Fig 6A of Sack. This shows the binding of dihydrotestosterone to the AR ligand-binding domain.
- It is common ground that the skilled reader would understand that the best way to read the Poster is to start in the middle as the document shows a progression in the design of small molecule (non-steroidal) inhibitors according to the direction of the arrows around the page. SAR studies are shown on the right, followed by PK-DM optimisation on the left.
- Following the upward arrow in the centre of the page shows that the authors had progressed from the starting compounds (nilutamide and RU59063) to RD2 and then through various further “RD” compounds in a clockwise direction.
- The Skilled Medicinal Chemist would understand that in these studies the authors were seeking to optimise the structure by making rational modifications in a step wise fashion to investigate the impact on activity in the relevant prostate cancer models.
- The first compound is RD2, which the skilled medicinal chemist would note had structural similarities to nilutamide and in particular to RU59063 (the only difference being the change from a hydroxy group (OH) to an azide group (N3) on the right-hand side of the molecule.
- The authors then move to RD6 where they have introduced a phenyl group on the nitrogen. RD6 still contains the azide group but it now sits on the phenyl ring, rather than the alkyl chain in RD2.
- The next molecule is RD7 where the azide group has been replaced with a methyl group (of similar size to an azide but less reactive and which is also lipophilic).
- The difference between RD7 and RD37 is at the position X, with a dimethyl replaced with a cyclobutyl group. This can be characterised as a small chemical change with the introduction of an additional CH2 group.
- The final compound in this section is RD54 which has two differences over RD37:
i) First, the authors have further expanded the size of that bottom ring (at position X) to a cyclopentyl. The skilled medicinal chemist would understand the authors had made these modifications in RD37 and RD54 to investigate the impact of different sized groups in the binding pocket. It is apparent that the dimethyl, cyclobutyl and cyclopentyl groups at this position are all consistent with antagonist activity.
ii) Second, the methyl group on the phenyl ring (on the right) has been replaced by a cyano group (CN).
- The other half of the page (on the left-hand side) is labelled PK-DM Optimisation and features three RD compounds, RD131, RD161 and RD162.
- In RD131, the left-hand side and centre are the same as in RD37 (including the cyclobutyl group at position X). The authors have started to modify the right-hand ring and have introduced new substituents at the 4-position, specifically an N-methylbutyramide group (i.e. a methyl amide linked to the phenyl group via a propyl chain).
- In RD161, the methylamide group is directly attached to the phenyl ring, and the Skilled Medicinal Chemist would note this would make the compound more rigid.
- In RD162 (shown in a different colour to the other compounds) the authors have added a fluorine on the phenyl ring. The Skilled Medicinal Chemist would consider this had likely been done to improve metabolic stability compared to RD161 (although the PK data for RD161 is not shown), since s/he would know that the phenyl ring might be susceptible to oxidation, a precursor to elimination of the drug metabolite and a measure of instability, and adding a fluorine was a common approach to preventing metabolic oxidation.
- From those changes, the Claimants drew attention to the following particular points:
i) RD7 and RD37 differ only in that the former has a geminal dimethyl group at the bottom right of the central ring, whereas the latter has a spiro cyclobutyl group in that position.
ii) RD54 has a cyclopentyl group in that position, but also has a cyano (CN) group in place of the methyl (CH3) on the top right position of the right-hand aromatic ring.
iii) RD161 and RD162 differ only in that the latter has a fluoro group on the right-hand aromatic ring. Prof Westwell’s evidence is that the Skilled Medicinal Chemist would consider that this addition had likely been done to improve metabolic stability compared to RD161, albeit that PK data for RD161 is not shown (see below). Profs Ward and Westwell agree that adding a fluorine was a common approach to preventing metabolic oxidation.
The cell-based data
- The chart at the top right shows data from an antagonist assay on hormone sensitive (HS) prostate cancer (“HSPC”), and the chart at the bottom shows data from an antagonist assay on hormone refractory (HR) prostate cancer. These would both be understood to be cell-based assays, which are measuring relative PSA (prostate-specific antigen) levels and assessing the dose response of each of the compounds. PSA expression is being used as a surrogate for cell growth. A lower value for the relative PSA level indicates higher antagonist activity.
- The HS assay data (reproduced above) shows bicalutamide (‘Bic’) reducing relative PSA level in a dose-dependent manner i.e. it is behaving as an antagonist as expected in HSPC. It also shows a dose response for both RD2 and RD6, with at least RD6 performing better than bicalutamide.
- The HR assay data (reproduced above) shows that bicalutamide (‘Bic’) is not reducing PSA levels, indicating that it is not behaving as an antagonist in HRPC. However, all the RD compounds show much improved antagonist profiles and more significant reduction of PSA levels (particularly from RD6 onwards).
The in vivo data
- In the centre on the right of the Poster is data from an in vivo assay which measures change in tumour volume over time following administration of bicalutamide, RD7 and RD37 over a number of days. A smaller increase in tumour size over the same period would be understood to show higher activity of a compound. The reference to “LNCaP/HR” would be understood to be a reference to the cell line that was used, with “HR” standing for hormone refractory. It would be understood that the experiment likely involved implantation of cells from this human cell line into an animal (immunocompromised mouse) model in order to measure tumour growth in vivo.
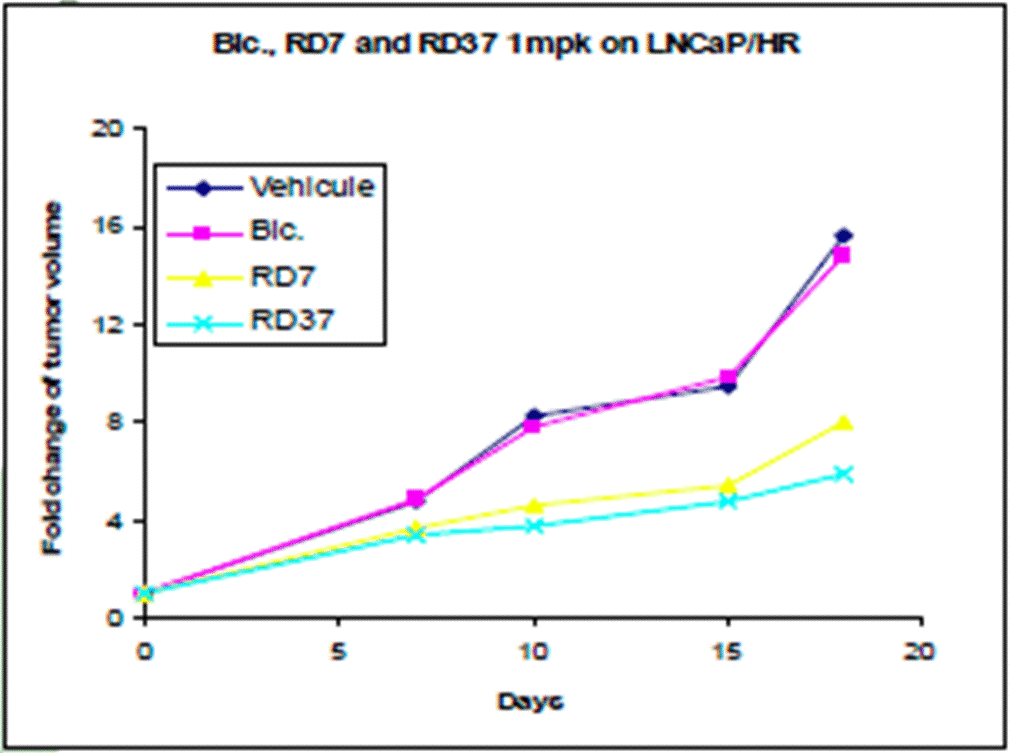
- The data show that bicalutamide does not have an effect on HR tumour growth compared to the vehicle. In contrast, RD7 and RD37 have a discernible impact, both slowing tumour growth at a similar rate. The extent to which any meaningful distinction can be drawn between the performance of RD7 vs RD37 (i.e. the blue vs yellow lines) was a point in dispute which I address later.
The PK data
- The left-hand side of the Poster relates to ‘PK-DM optimisation’. ‘PK-DM’ means “pharmacokinetics drug metabolism” which broadly relates to how a compound behaves in the body. Three RD compounds are shown on this side: RD131, RD161 and RD162.
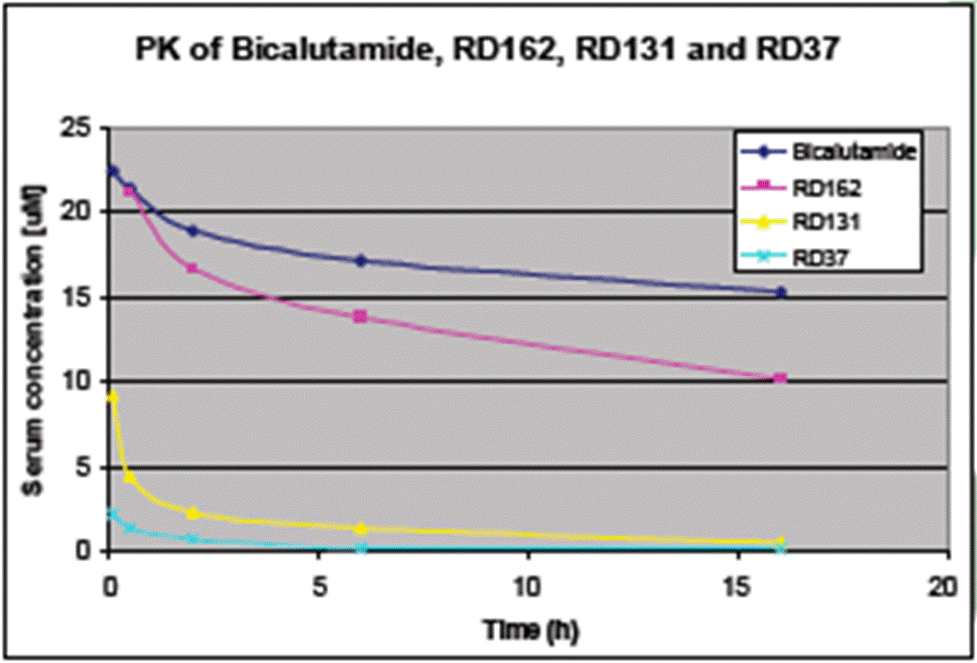
- The chart on the middle of the left-hand side (reproduced above) shows that the serum concentration of RD37 and RD131 drops off very quickly after administration whereas RD162 takes longer to clear and has a profile closer to that of bicalutamide. It is common ground that the PK profiles for RD131 and RD37 would be off-putting for once daily oral dosing, whereas that of RD162 is significantly better.
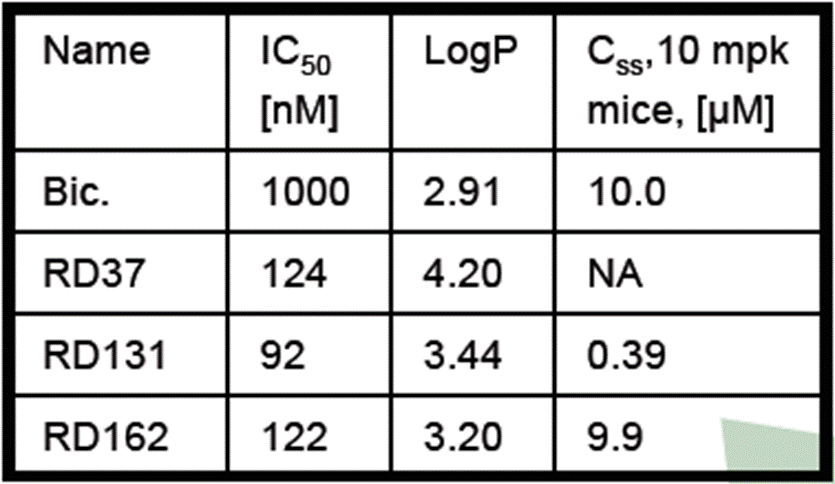
- The table at the top of the left-hand side of the Poster (above) compares the IC50, LogP and steady-state concentration of RD37, RD131 and RD162. The following was common ground:
i) Compared to bicalutamide, all three RD compounds are more potent antagonists (shown by their lower IC50 values).
ii) The IC50 values for the three RD compounds are approximately the same.
iii) Of the three RD compounds, RD37 is the most lipophilic (highest LogP), whilst RD162 is the least lipophilic (lowest LogP). Of the three RD compounds, the LogP value of RD162 is the most credible for a drug candidate.
iv) RD162 has a significantly better steady-state concentration than RD37 and RD131 and is comparable to bicalutamide, which is consistent with the data in the PK chart.
- Prof Westwell was of the view that, having reviewed the data presented, the Skilled Medicinal Chemist would understand the following from the Poster:
i) Based on the IC50 values, all the RD compounds were more potent inhibitors of the AR than bicalutamide.
ii) RD162 had been shown to have better therapeutic potential than RD37 and RD131 based on its improved PK-DM properties.
iii) Even though there are no PK data shown for RD161, the implication of the arrow pointing to RD162 and it being highlighted in blue, is that RD162 had improved PK-DM properties compared with RD161.
iv) Overall, what the authors summarised in the Poster was the identification of a compound RD162 which had the potential for therapeutic activity as an AR antagonist in HRPC and potential advantages over the other compounds tested because it demonstrated acceptable PK-DM properties.
- Furthermore, although there is no data for RD162 in the HS model, the Skilled Medicinal Chemist would assume that RD162 had potential for therapeutic activity in HSPC. RD2 and RD6 were shown to be active in the HS assay and the HR assay. Given that all the RD compounds were suggested to be AR antagonists, the Skilled Medicinal Chemist would expect the RD compounds that follow those also to show activity in the HS assay. Therefore, the Skilled Medicinal Chemist would assume that RD162 would be active in both assays.
- In the parties’ opening skeleton arguments, my attention was drawn to familiar authority. However, in view of the critical importance of obviousness in this case, I will set out the parties’ contentions to ensure I have them well in mind.
- Naturally both sides referred me to the principles regarding classical obviousness as reviewed by the Supreme Court in Actavis v ICOS [2019] UKSC 15.
- At [57] Lord Hodge explained that the general principle of ensuring that the extent of monopoly corresponds to the technical contribution of the patent permeates all aspects of validity:
57. The general principle that the extent of the patent monopoly should correspond to and be justified by the actual technical contribution to the art is thus part of the jurisprudence of both the EPO and the UK courts and, as Lord Sumption observed in Generics v Warner-Lambert (above), para 17, "the principal conditions of validity, novelty, inventive step, industrial application and sufficiency are all, in one way or another, directed to satisfying the principle thus expressed".
- It was also reiterated that the statutory question is whether the invention is obvious, having regard to the state of the art at the priority date. In some cases, it is helpful to answer this question by adopting the structured approach set out in Pozzoli. In other cases, it is helpful to adopt the problem/solution approach (PSA) favoured by the EPO. But neither approach can replace the statutory question itself. It must be assessed by reference to the facts and circumstances of the case. See [58]-[62].
- At [63] Lord Hodge cited Kitchin J (as he then was) in Generics (UK) Ltd v H Lundbeck [2007] EWHC 1040 (Pat), [2007] RPC 32 emphasising the importance of the facts and presenting a list of illustrative, but not exhaustive, factors to take into account (emphasis added):
63. In Conor Medsystems Inc v Angiotech Pharmaceuticals Inc [2008] UKHL 49; [2008] RPC 28; [2008] 4 All ER 621, at para 42 Lord Hoffmann endorsed the fact-specific approach which Kitchin J set out in Generics (UK) Ltd v H Lundbeck [2007] RPC 32, para 72 where he stated:
"The question of obviousness must be considered on the facts of each case. The court must consider the weight to be attached to any particular factor in the light of all the relevant circumstances. These may include such matters as the motive to find a solution to the problem the patent addresses, the number and extent of the possible avenues of research, the effort involved in pursuing them and the expectation of success."
Kitchin J's list of factors is illustrative and not exhaustive. Another factor which needs to be considered in the present case is the routineness of the research. Much of the interest and controversy which the Court of Appeal's judgment has generated relates to how people have understood or misunderstood the significance which that court has attached to the routine nature of the pre-clinical and clinical research which I have described.
- Whether the inventive step would have been “obvious to try” is a question that has been considered in many cases. In Actavis v ICOS at [65] Lord Hodge stated:
65. First, it is relevant to consider whether at the priority date something was “obvious to try”, in other words whether it was obvious to undertake a specific piece of research which had a reasonable or fair prospect of success: Conor v Angiotech (above) para 42 per Lord Hoffmann; MedImmune Ltd v Novartis Pharmaceuticals UK Ltd [2012] EWCA Civ 1234; [2013] RPC 27, paras 90 and 91 per Kitchin LJ. In many cases the consideration that there is a likelihood of success which is sufficient to warrant an actual trial is an important pointer to obviousness. But as Kitchin LJ said in Novartis AG v Generics (UK) Ltd [2012] EWCA Civ 1623, para 55, there is no requirement that it is manifest that a test ought to work; that would impose a straightjacket which would preclude a finding of obviousness in a case where the results of an entirely routine test are unpredictable. As Birss J observed in this case (para 276), some experiments which are undertaken without any particular expectation as to result are obvious. The relevance of the “obvious to try” consideration and its weight when balanced against other relevant considerations depend on the particular facts of the case.
- There can be multiple obvious avenues or routes and an obvious route is not rendered less obvious for this reason. At [69] in Actavis v ICOS the Supreme Court approved the well-known passage from Brugger v Medic-Aid at 661:
“[I]f a particular route is an obvious one to take or try, it is not rendered any less obvious from a technical point of view merely because there are a number, and perhaps a large number, of other obvious routes as well.”
- Lord Hodge noted his agreement and added that “[a]s a result, the need to make value judgments on how to proceed in the course of a research programme is not necessarily a pointer against obviousness.”
- Further, the Supreme Court in Actavis v ICOS was clear that if a Skilled Team engages in familiar and routine testing and it is obvious to undertake that testing as part of routine development, that is sufficient for obviousness. The end result of those routine tests does not need to be known or to be anticipated or expected. It is obtained by the application of routine work (see for instance [85] and [88]).
- The Claimants submitted that three particular issues that arise in this case concern Astellas’ arguments that (1) there was no motivation to make any change to the prior art, (2) there was a perceived prejudice against taking a particular step (namely, of including a geminal dimethyl group) and (3) the irrelevance of ‘a golden bonus’. I address these points in turn.
- Motivation was a key issue identified by both sides. For their part, the Claimants submitted that the following points of principle need to be kept in mind, and in this section, all the emphasis in the cited passages is that of the Claimants.
- First, the correct way to frame any question about motivation is in terms of whether the skilled person “would have a motive to find a solution to the problem the patent addresses”. See Lord Hodge in Actavis v ICOS at [63], which is cited above. It is wrong to jump straight to the question of whether “the skilled person would have a motivation to change the prior art” until one has decided what the problem is that the patent is addressing. That is important in this case because, the Claimants submit, the Patent is not addressing the problem of making a better compound than RD162, merely an alternative.
- Second, the reason it is often wrong to pose any question about motivation in terms of whether there is a motivation to change the prior art is because doing so risks putting the notional skilled person into the shoes of the author of the prior art and thereby confounding the question of obviousness with irrelevant considerations. See Laddie J in Brugger v Medic-Aid Ltd [1996] R.P.C. 635 at 655:
The plaintiffs' case was that there had been no sea-change between 1966 and 1984 in what nebulizer manufacturers were trying to achieve by way of performance characteristics of their products and that the existing nebulizers made in accordance with '355 were highly regarded. In particular they said:
"There was no evidence that as at 1984 there was an identified problem to solve with the '355 (or the '920). In particular it was not shown that there was an identified problem with the '355 that it had too low a proportion of respirable particles. The only contemporary evidence is the Warentest published in Germany only which showed that the '355 device had a higher RAO than the Heyer device which was the nearest competitor. More generally, although there may have been some increase in competition in the market in the 1980s, the plaintiffs' nebulizers were better than the competition. Professor Balachandran accepted that what the skilled man would do by way of attempting to improve '355 would depend on what the commercial brief was."
On the basis of this they say there is no reason why it should have been obvious to modify the prior art in any particular direction. That, it appears to me, is a non sequitur. The fact, if it be one, that existing commercial products are highly successful and satisfactory does not indicate that there are no obvious modifications to make to them. It merely demonstrates that there may be little incentive to those already making those products to change the design - a quite different matter.
- On this point, the Claimants also referred to this passage from the judgment of Mr Daniel Alexander QC in Meter-Tech Llc & Anor v British Gas Trading Ltd (Rev 1) [2016] EWHC 2278 (Pat) at [325]:
325. The issue of mindset, in so far as it enters the picture, is one aspect of an overall evaluation of whether there was or was not a motivation or reason for the skilled person to take a given step. Motivation can of course be important in evaluations of obviousness (see for example Hoechst Celanese v. BP Chemicals [1997] FSR 547 – "necessary to demonstrate that there is some reason for taking [the step from the prior art]" and Lundbeck above). However, again here, it is dangerous to make too much of it as an isolated factor, because a product can be obvious even if there was no reason actually to make it at the priority date and a skilled person would believe it to be unusable (a 100cm dinner plate, to adapt a familiar example). It is also important to bear in mind that motivation can lie in the wish to make a different product which operates in a similar way, not necessarily a better one.
- Third, that is not to say that commercial considerations that the Skilled Team may have are completely irrelevant. As Lewison J observed in Ivax v Akzo Nobel BV (No.2) [2007] RPC 3 at [47]:
Commercial reasons may, in my judgment, be sufficient reason to take a skilled person down a particular path. I see no reason why those commercial reasons should exclude the desire to emulate a successful product, without infringing a patent, if the means of doing so are technically obvious.
- The Claimants also submitted that it has also been observed that an important influence that encourages the making of analogues of prior art compounds is “a desire to obtain a novel, as well as effective, compound” and that “the requirement of novelty for the grant of a patent provides a powerful driving force to investigations of this kind”. See Pumfrey J in Monsanto v Merck [2000] RPC 709 at [169]. The Claimants made this point in view of some evidence they adduced under their CEA notice as to what motivated the inventors to go beyond what was disclosed in the prior art.
- Fourth, motivation to take a particular step is, in any event, not a necessary condition to a finding of obviousness. See Floyd J. in Research In Motion UK Ltd v Visto Corp [2008] EWHC 335 (Pat):
“73. A question which often arises, and arises here, is whether the absence of a particular motive to take a particular step between the prior art and the invention is fatal to an obviousness attack. It must now be regarded as settled law that it is not. In Pharmacia v Merck [2001] EWCA Civ 1610; [2002] RPC 41 , Aldous L.J. cited with approval a passage in Laddie J.'s judgment in Hoechst Celanese v BP Chemicals [1997] FSR 547 at 573. The court will readily assume that technicians and businessmen will wish to make trivial changes to what is known in order to produce essentially the same result. That is not to say motive is irrelevant: it is one of the many factors that has to be balanced in answering the statutory question. It is easier to show a step is obvious if there is a strong motivation to take it. If the advance is not as trivial as the Pharmacia and Hoechst cases require, the absence of motivation is a factor of which account must be taken in the balancing exercise.”
…
112. RIM accepts that there was no motive, in this scenario, to make the change. But it submits that that does not matter for Pharmacia / Hoechst reasons. The modified system is nevertheless obvious technically: it does essentially the same thing in a different way. The technique of passing data through a firewall using HTTP was a well known tool. Why should anyone not be free to use it if the need ever arose?
113. Visto's rejoinder is this: it is not correct that HTTP tunnelling does the same thing. The use of HTTP tunnelling solves a technical problem: how one gets through the firewall without having to reconfigure it. So the Pharmacia / Hoechst reasoning does not apply.
114. I think these difficulties are created by fixing on a starting point where it has already been decided how the e-mail is to be transported through the firewall. If one has already decided to do something in a particular way, it will be seldom to be a logical step to retrace one's steps and do it in another way, particularly if that alternative way provides no advantage, as it would not in the case of a licensed Lotus Notes user. To say to a Lotus Notes user who has already configured his firewall that a different port is available would not solve any technical problem with which he is faced or provide him with any technical benefit. To use the familiar example: if a piece of wood is satisfactorily screwed to another, there is no reason to take it apart and glue it instead; but it is certainly not inventive to do so. And it does not make it inventive if you say that doing so avoids the technical problem of how to fix together two pieces of wood without using screws.
- In terms of the applicable principles, I accept the Claimants’ submission that a patentee cannot rely upon a perceived problem in taking a particular course of action in support of inventive step, unless the patent itself overcomes that problem, based on Philips v Asustek [2019] EWCA Civ 2230 at [73], and Jacob LJ in Pozzoli v BDMO [2007] EWCA Civ 588 at [28]. The Claimants’ submission was that, in this case, if there was any perceived problem associated with geminal dimethyl groups, that problem would be just as apparent having read the Patent as having read the prior art.
- The applicable principle is best explained in the context where it is said to arise. The Claimants make this point in response to any suggestion from Astellas that the skilled person would not expect RD162’ to be a ‘superior’ compound to RD162, and they say this does not assist the Defendants. The Claimants’ position is that it would have been obvious to the Skilled Team that replacing the cyclobutyl group at position X of RD162 with a gem-dimethyl moiety would likely give rise to an active compound of similar potential to RD162. Even if (which is not accepted) RD162’ turned out to be a ‘superior’ compound that does not help if it was obvious to make it anyway. See e.g. Lord Hodge in Actavis v ICOS at [73] (a “golden bonus” does not found a valid patent if the claimed invention was obvious for another purpose), and Napp v ratiopharm [2009] EWCA Civ 252 at [115].
- For their part, Astellas particularly stressed the well-known passage from Kitchin J in Generics v Lundbeck (quoted in [181] above). Astellas also emphasised that motivation (a) almost always plays a part in the overall assessment of obviousness and (b) is an important tool to avoid a hindsight approach. Furthermore, as they submitted, it is settled law that the Skilled Team does not undertake technical tasks for the sake of doing so or out of idle curiosity (citing Lord Hodge in ICOS and the TBA in AgrEvo, as quoted by Lord Hodge at [70]).
- In both their opening and closing submissions, Astellas addressed a number of submissions directed to their perception that the Claimants’ case was that the change from RD162 to RD162’ was trivial and obviously so. These comprised a series of reminders from the relevant case law which I summarise as follows.
- First, I agree that there can be real danger in saying that just because a modification is “small” or “conservative” in structural terms, that it is obviously “trivial” and will obviously have no material effect. Chemical structures are very different from plate diameters.
- Second, a case run on “obvious immateriality” does not absolve the Claimants from having to consider the actual teaching of the prior art or allow them to generalise it into oblivion. It is also important not to work backwards from the solution as disclosed in the Patent. The test remains whether the Patent is obvious over the cited prior art teaching. Floyd LJ stated in Philips v Asustek [2019] EWCA Civ 2230; [2020] RPC 1 at [61] that the Court does not approach obviousness by taking the prior art and “stripping out from it detail which the skilled person would otherwise have taken into account, or ignoring paths down which the skilled person would probably be led”.
- Although there is no attack based on CGK in this case, it is nonetheless relevant that the “generalised to oblivion” approach is why an approach based on the CGK alone can be so unfair on patentees. As Floyd J (as he then was) explained in ratiopharm v Napp [2008] EWHC 3070 (Pat); [2009] RPC 11 at [158], the danger is that the starting point is not “obviously encumbered with inconvenient details of the kind found in documentary disclosures”.
- Third, such an approach cannot be used to try to conceal or dodge a lack of any motive to take a step or that the prior art might teach away from it. Motive almost always has a role to play in any obviousness analysis. In Napp v ratiopharm [2009] EWCA Civ 252; [2009] RPC 18, the difference between the claimed invention and the prior art was a slower release formulation. The obviousness case was rejected by the Court, with Jacob LJ explaining at [111] that “Unless there was a point in pressing on, why should the skilled person embark on what would be a somewhat troublesome, even if uninventive development programme? The answer is the same as for the case on common general knowledge - there was no point”. The Judge explained at [113]: “It makes no sense to say that a formulator would press on and develop a 12-hour release formulation without any apparent clinical point. Motive remains relevant on any view.”
- This is mirrored by the Supreme Court in Actavis v ICOS where it was explained (at [70]) that “the motive of the skilled person is a relevant consideration. The notional skilled person is not assumed to undertake technical trials for the sake of doing so but rather because he or she has some end in mind”.
- As Kitchin J (as he then was) explained in the passage from Generics v Lundbeck quoted above, motivation is usually important. There are some cases where motivation may play a lesser role or is not determinative, for instance where the change from the prior art is arbitrary or entirely trivial (and where the prior art does not teach away or is neutral), meaning that the need for motivation to take a particular step might not play a part in the analysis. Jacob LJ used the example of changing the diameter of a plate by ¼ inch, but such cases are rare, and Astellas submitted, particularly so in a chemical or pharmaceutical context.
- Finally, I was reminded (again) that care must be taken in approaching the Pozzoli questions to avoid hindsight. The statutory question is always whether the claimed invention is obvious without knowledge of the Patent or the claimed invention.
- In their closing, Astellas responded to the case law cited by the Claimants in their Opening on this topic, as follows.
- Once again, Astellas warned that great care must be taken in seeking to apply case law on obviously trivial changes to a pharmaceutical context. In Hoechst Celanese v BP Chemicals [1997] FSR 547 at [573], Laddie J had used the phrase “workshop modification” and suggested that almost no motivation is needed to make such a change. The Court of Appeal in Pharmacia v Merck did not agree that it was appropriate to refer to “workshop modifications” as this was introducing a test not in the statute: the statutory test is obviousness (at [124]).
- The Claimants in the present case rely on RIM v Visto. The invention in RIM v Visto concerned how to send and synchronise emails from outside and inside a firewall. In that case, the invention used an HTTP communication, and the prior art Lotus Notes application used RPC (through a different port) rather than HTTP (see Judgment at [65] and [106]).
- The argument was that there was no reason to change Lotus Notes as it worked perfectly well crossing the firewall using RPC through port 1352, there was no reason to use HTTP through port 80. It was in that context that the Judge considered the case law on modifications which do essentially the same thing in a different way and therefore need little or no motivation to do (see at [111]-[121]).
- In RIM v Visto, it was CGK that HTTP could be used to transfer data (and that it used port 80) and that it was a “generic request/response protocol that can be used for a variety of tasks” (at [14]). The distinction was to change one CGK protocol to transfer emails with another CGK protocol for doing the same thing. That took no invention and required no motivation to do: it could just be done without any concern and without any uncertainty as to the outcome.
- The problems in applying such an approach to a case of chemical substitution on medicinal compounds are plain. If such an approach is to be applied, it truly must be obvious that the change will make no material difference (i.e. it is obvious that the components at issue can be swapped without concern or impact). That was the case in RIM v Visto and is the case when considering diameters of plates or different fixing means. Astellas submitted that this point simply does not apply in the present case.
- Astellas submitted that the fallacy in the present case lies in the proposition that a seemingly small structural change is tantamount to an obviously immaterial one. The one does not follow from the other in medicinal chemistry.
- These points were aimed by Astellas at the ‘obvious to do SAR’ case.
- In ICOS the issue arose over obviousness of routine tests. Lord Hodge made the following observations which are important to keep in mind:
i) The routine nature of the research and any “established practice of following such research through to a particular point” may be a relevant consideration [66].
ii) However, it should also be remembered that the “need to facilitate expensive pharmaceutical research is an important policy consideration for legislators and others involved in intellectual property law” [67].
iii) Although a number of routes may each be obvious, “the existence of alternative or multiple paths of research will often be an indicator that the invention contained in the claim or claims was not obvious” [69].
iv) Motive is a relevant consideration. The notional skilled person is “not assumed to undertake technical trials for the sake of doing so but rather because he or she has some end in mind” [70].
v) It is not sufficient that a skilled person could undertake a particular trial; “one may wish to ask whether in the circumstances he or she would be motivated to do so. The absence of a motive to take the allegedly inventive step makes an argument of obviousness more difficult” [70].
vi) The fact that the results of the research which the inventor actually carried out are unexpected or surprising is a relevant consideration, at least in so far as it suggests that a test was not obvious to try or otherwise in the absence of a known target of the research which would make it less likely that the skilled person would conduct a test [71].
vii) Care must be taken in respect of a “step by step” analysis of what a skilled person might do unless the steps can be ascertained without the taint of hindsight [72].
- In ICOS, the claim was to a maximum of 5mg tadalafil per day. The Judge had found that the Skilled Team would investigate the lowest dosage possible under a routine dosage study but would not have known the 5mg in advance. In the Court of Appeal and Supreme Court it was held that this did not matter: the dosage would be found in any event by entirely routine testing which was obvious (indeed necessary) to undertake.
- The ICOS judgment cannot be used to advance the proposition that just because embarking on testing is routinely done or even “bread and butter” in nature, the results are thereby obvious. That would be contrary to the important public policy of supporting pharmaceutical research.
- The point about the dose ranging studies in ICOS is that they were always done to seek to obtain the lowest dose possible consistent with effectiveness, whatever that dose might be. Lord Hodge explained at [77] that “The target of the skilled person’s research is in large measure pre-determined” (emphasis added). This is because, for any drug, the lowest effective dose will exist and it is simply a pre-determined matter of lowering the dose successively to identify it.
- The key to the finding in ICOS was that the routine testing was obvious to do and the end result was in large part pre-determined (all that was left was to put a number on the minimum effective dose). As Lord Hodge said at [88]:
“Standing back from the step by step analysis, it is clear that the skilled team was engaged in the familiar and routine testing of a drug to establish the appropriate dosage regime for tadalafil in order to implement the teaching of the Daugan patent. The target was never in doubt. It was obvious to embark on that exercise and carry out tests in a routine way until that appropriate dose was ascertained.” (emphasis added).
- In opening, the Claimants placed considerable emphasis on the judgment of Pumfrey J (as he then was) in Monsanto v Merck [JA/2] concerning COX II inhibitors (see their opening skeleton at §106 and T1/2920-3223). No wider propositions of law are relied upon that are said to arise from this case. Astellas suggest that the Claimants rely on it because it is a case where it was held there was motivation to do a SAR and that the patent was obvious as a result. Astellas also submitted that the dangers of seeking to draw wider propositions from something so factual are plain. Indeed, what was not pointed out in opening was how different the facts of Monsanto were to the present case.
- In Monsanto, the prior art “Gans” was a paper that disclosed the anti-inflammatory activity of the compound DuP 697 in rats and stated it did not produce intestinal or gastric ulcers (the problem with non-steroidal anti-inflammatory drugs). DuP 697 was the 2,3 isomer of the claimed compound (the 3,4 isomer).
- The Judge accepted that it would be obvious to undertake a routine SAR and find the 3,4 isomer of the claim. But the evidence was telling. Not only was DuP 697 the focus of Gans (and so the starting point was not in doubt) but both the Claimant and the Defendant in that case had independently seen DuP 697 and both had quickly synthesised the 3,4 isomer. It is perhaps not surprising that the Judge held that the 3,4-diaryl substitution would be “one of the first things which would occur to the medicinal chemist” (see at [159] and [164]-[172]).
- This was not a case where just “doing a SAR” of some unknown scope and aim was held to be obvious. It was a case where the starting compound was not in doubt and where both parties had actually themselves started from it and both got to the same place quickly and which on the evidence the Judge held would be one of the first things that would be done to the disclosed compound.
- Astellas contended the present case is very different indeed.
- Finally, Astellas emphasised that it is impermissible to seek to mix up what is said to be the technical contribution over a piece of prior art and the question of obviousness.
- In Conor v Angiotech, it was argued that obviousness should be assessed based on a watered-down version of the alleged invention (said to be based on what was the actual contribution of the patent over the cited prior art), rather than by the claimed invention. This was rejected. Lord Hoffmann explained (at [15]-[17]) that this type of argument is an “illegitimate amalgam of the requirements of inventiveness (Art. 56 of the EPC) and either sufficiency (Art. 83) or support (Art. 84) or both”. Lord Hoffmann explained that it is the claimed invention which has to involve an inventive step. The invention means prima facie that specified in the claim: see s.125(1) of the 1977 Act.
- Finally, under this heading, Astellas made the well-known point that the actual teaching of the prior art (as a whole) also cannot be ignored, relying on the dictum of Floyd LJ in Asustek which I set out at [199] above.
- I have endeavoured to keep all these principles in mind.
- Although it is trite that the Poster and the Slides must be considered separately (since, as I understand matters, the Claimants abandoned in advance of trial their case that the Poster and the Slides would be read together by the Skilled Team), much of the reasoning and the attacks related to both. Nonetheless the disclosures are not the same and it is important not to elide the two, so I consider the case over the Poster first.
- The Claimants submitted in opening that their case on obviousness can be stated very simply as follows.
- At the relevant date there was a motivation to find treatments for HRPC. Applying Pozzoli:
i) The inventive concept of the Patent is that a compound with the structure of RD162’ has potential for treating prostate cancer or HRPC.
ii) The difference between that and the Poster is that the compound of claim 1 (RD162’) has a dimethyl substituent at position X of the central ring whereas RD162 has a cyclobutyl substituent at that position;
iii) It would have been obvious to the Skilled Team that compounds with a similar structure to RD162 are likely to have similar activity, in particular it would have been obvious that a compound which is the same as RD162 but has a cyclopentyl or dimethyl substituent at position X would be likely to exhibit similar activity against hormone refractory cells.
iv) In the case of [the Poster], the Skilled Medicinal Chemist is taught that compounds having any of a dimethyl, cyclobutyl or cyclopentyl group at the bottom right position of the central ring have good in vitro activity (RD7, RD37 and RD54).
v) From that starting point, it would therefore have been obvious to the Skilled Medicinal Chemist that these specific variants of RD162 (including RD162’) would be likely to have the same or similar activity, and therefore also have therapeutic potential to treat prostate cancer.
- The Claimants’ closing was slightly more nuanced - indeed Astellas say the case was different. So far as the Poster was concerned, the Claimants emphasised the following ‘key teachings’:
i) All the compounds have, on the left side, a phenyl ring with a trifluoro and cyano group. The same group was also present in RU59063.
ii) The phenyl ring on the right side of the molecule was associated with improved in vitro activity against hormone refractory prostate cancer cells. Improved activity meaning more active than bicalutamide (and more active than RD2).
iii) RD37 and RD131 are not suitable due to poor PKDM properties.
iv) The introduction of a methyl amide at position 4 on the right phenyl ring and combined with a fluorine (F) at position 3 produces acceptable PK properties.
v) At position X the inclusion of gem-dimethyl, cyclobutyl and cyclopentyl were all consistent with improved in vitro activity (over bicalutamide and RD2).
vi) There is a pairwise comparison between RD7 and RD37 which did not demonstrate a material difference in activity between the dimethyl and the cyclobutyl.
- On that basis, the Claimants submitted that it was (i) obvious that replacing the cyclobutyl in RD162 with gem-dimethyl would make a compound which is likely to have therapeutic potential as measured by an in vitro assay and/or (ii) it was obvious to test such a compound in an in vitro assay for that purpose, in which case it would be shown that it would have therapeutic potential.
- I will start by addressing the Claimants’ primary evidence on obviousness over the Poster, together with the attacks on that evidence, before considering Astellas’ positive case of inventiveness.
- Prof Westwell’s key reasoning regarding the Poster was set out in his first report as follows:
‘9.49 From the disclosure in [the Poster] the skilled medicinal chemist would expect certain variants of RD162 to have the same or similar activity and therefore also have therapeutic potential. In particular, the skilled medicinal chemist knows from the teaching in [the Poster] that the cyclobutyl at the bottom right position can be substituted with a dimethyl or a cyclopentyl (as shown in RD7 and RD54) without having a significant impact on in vitro activity.
9.50 [The Poster] shows that the most significant development in achieving in vivo serum stability (at a comparable level to SOC bicalutamide) is the installation of a 3-fluoro substituent in the aryl ring joined to the central thiohydantoin, which the skilled medicinal chemist would understand to be a common method to prevent liver-based cytochrome P450 metabolism for orally administered drugs, as discussed. Therefore, although predicting the site of metabolic oxidation is often difficult based on chemical structures alone, the skilled medicinal chemist would understand that the aryl ring joined to the central thiohydantoin was the likely site of metabolic oxidation because RD162 (which includes the 3-fluoro substituent in the aryl ring) is shown to be stable. There is no reason to consider that a change from the cyclobutyl group to either a dimethyl or cyclopentyl would affect metabolic stability. This is because these are small changes which retain the same hydrocarbon functionality and are made at a site that is remote from the metabolic site. Additionally, the routine incorporation or removal of a single CH2 group to this hydrophobic part of the molecule (going from dimethyl to cyclobutyl or vice versa) would not significantly affect properties such as logP and consequently would not change properties such as solubility or in vivo biodistribution, in any significant way.
9.51 It would therefore be immediately obvious to the skilled team that the following two compounds would be likely to have therapeutic potential that was similar to that of RD162.’
[Emphasis added. Underneath this paragraph, Prof Westwell showed RD162, and two RD162 analogues - one where the cyclobutyl was replaced with dimethyl and the second where it was replaced with a cyclopentyl group].
- Having identified that the Teutsch paper (referenced in the Poster) gave a general synthetic route to the core arylhydantoin structure of the RD series, Prof Westwell said in [9.52] that through appropriate choice of commercially available starting materials, that method could be extended to provide the RD series structures.
- Prof Westwell then went on to consider changes to the other parts of RD162. In [9.53] he said this (with my emphasis added):
‘I have been asked to consider whether changes to other parts of RD162 would also result in molecules which would be expected to have similar therapeutic potential. Other changes could be investigated but the activity of such molecules prior to testing would be more uncertain ‘and therefore their therapeutic potential as compared to RD162 would not be as immediately obvious, in particular because of the potential for disadvantageous PK-DM properties compared to RD162.’
- The possible changes which Prof Westwell considered were:
i) The left-hand side. He pointed out that that part of the structure had been kept constant all the way from RU59063, and commented that it would be uncertain, based on the Poster, whether changes to that part of the molecule would be consistent with activity.
ii) Modifications to the top-right ring, but he indicated that the Skilled Medicinal Chemist would understand they could result in different properties, giving the examples that the addition of the N-methylbutyramide in RD131 did not have desirable PK properties and the methyl group in the same position in RD37 results in a compound with a higher logP and potential issues with drug solubility and steady state concentration.
iii) The 3-fluoro substituent. The inclusion of this fluorine would be understood as likely to improve metabolic stability and would be retained for that reason. The Professor considered the possibility of moving it to the 2-position (equivalent to the 5-position) but indicated the effect would be uncertain since although fluorine atoms are commonly used to resolve issues of metabolic stability, their effect is unpredictable, and the move could have a negative effect.
- Before I return to consider the attacks on the Claimants’ evidence, it is convenient first to consider the three reasons which Astellas put forward as to why they said RD162’ was inventive. Each was based on evidence given by Prof Ward. In summary these were:
i) ‘there was no motivation for the skilled chemist to take up or continue with any medicinal chemistry work on any of the RD series of compounds’.
ii) The Skilled Medicinal Chemist would have a concern that ‘a dimethyl substitution would typically be viewed as less metabolically stable’ and therefore s/he would not consider RD162’ obvious.
iii) There was a hydrophobic pocket which would be expected to fit the cyclobutyl better than the dimethyl because of its additional bulk, and better than the cyclopentyl (presumably because of its lesser bulk).
- The Claimants were dismissive of each of these three reasons, characterising each as, respectively, misguided, misleading and abandoned. These reasons were also the focus of the Claimants’ criticisms of Prof Ward. Accordingly, it is necessary to analyse the evidence and the arguments. I will first consider the criticisms of Prof Ward which were not directly related to the three reasons.
- An overarching point made by the Claimants was that Prof Ward had experience giving expert evidence in patent cases, but that cannot, of itself, be a criticism.
- I shall deal with the Claimants’ specific points relatively succinctly. The first point was that Prof Ward had failed to equip himself with the relevant CGK of the Skilled Medicinal Chemist working on HRPC. However, as he stated in his first report, he had read the technical primer and the draft CGK section in Prof Clarke’s report, indeed the whole of his draft. He read the Chen, Marhefka, Sack and Teutsch papers and made various comments on the relevant content. The only part of his evidence where this criticism might have mattered was the suggestion that he did not have the experience to interpret biological assay data. He accepted he did not do the assays himself, but: ‘my job is interpreting assay data with the help of the biologist’. I reject this point.
- The second point was an accusation that he failed to answer questions put to him on three particular issues:
i) The first related to RD162’. There is no PK data for RD162’ in the Patent and the question which was put to him was whether it was plausible that RD162’ was suitable for use in therapy and whether this could be inferred from the structure of the molecule. The Claimants’ complaint was that this question had to be put at least 10 times. Prof Ward pointed out the data in Fig 21 and the fact that RD162’ had been placed in Tier 1 but said (repeatedly) that he would prefer to test before making any prediction. I have reviewed the whole passage of cross-examination T3/3163-32715. I do not consider Prof Ward was avoiding the question. The fact that the limited data in the Patent created something of an artificial situation for the Skilled Medicinal Chemist to consider does not mean there was a clear or easy answer to the questions being put.
ii) The second concerned his evidence about the metabolic stability of the gem-dimethyl group and whether there was anything in the Patent which would allay the skilled person’s fears on metabolic stability of that group. The criticism was that he refused to answer the question seven times. What actually happened in this passage of cross-examination was that Prof Ward immediately backtracked from his written evidence on this point (see further below), which meant there was nothing in the point.
iii) The third concerned a question put that in the absence of PK data in the Patent showing the relative performance of RD162, RD162’ and RD162”, it was not possible to tell which would be the best at treating prostate cancer. The criticism was that Prof Ward did not answer. His answer(s) was to point to the cell-based comparator data (in Fig 21) in conjunction with the interpretation of those data by the Skilled Cancer Biologist (i.e. Prof Clarke) and the Tier 1 ranking. It is difficult to know what more he could have said.
- The third allegation was that Prof Ward refused to accept propositions he had advanced himself. Again, three examples were relied upon:
i) The first concerned a passage in a paper by Lombardino and Lowe which he had quoted in his report as ‘aptly’ summarising the real-world medicinal chemist. It is true that he initially did not agree with it in cross-examination but when he had re-read the passage he agreed with it. This was nothing more significant than him not picking up the distinction between ‘a thorough knowledge of medicinal chemistry and an understanding of the biology’, when the passage was first raised.
ii) The second concerned this passage in a book chapter he had written in 2015 in Connolly & Ward (with the Claimants’ emphasis):
“Once a project has identified a compound that meets the candidate profile it becomes a possibility for nomination as a candidate drug. Usually, once one compound is found that fits the screening plan view of a candidate molecule, the project will be able to rapidly synthesise a range of close analogues that all fit, or come close to this standard. These compounds will all have subtly different properties. How does the project choose between them? One way is to assemble these compounds into a shortlist, and test all these compounds in a stricter fashion to define the best overall compound. At this point there are two main things to check before committing to extensive animal safety studies.”
iii) Prof Ward pointed out that this passage was in the very end section of a chapter on Lead Optimisation. His point was that the team would have developed synthetic expertise over the lifetime of their programme and would be able to make good progress synthetically towards the end of the programme. I think he also had in mind the knowledge obtained around the molecule from the SAR which would also have been conducted as part of the programme. He wanted to ensure the passage was considered in its correct context.
iv) I agree that this episode showed Prof Ward being a little cagey. Evidently, he could see what the cross-examiner was trying to get at which is why he was keen to explain the correct context. However, he was right to point out the context - the cross-examiner was trying to get an acceptance that, RD162 having been effectively presented on a plate in the Poster, the team would be able to rapidly synthesise a range of close analogues. That did not follow in the circumstances of the Poster.
v) There was also a minor related point where the Professor indicated this chapter had been revised in the next edition - a ‘totally new chapter’, yet this passage was the same in the 2023 edition. There is nothing suspicious about this: Prof Ward had a memory of a ‘totally new chapter’ but did not remember this passage was unchanged.
- The fourth allegation was that Prof Ward refused to answer questions on the issue of inventive step. The first point under this head concerned the assumption(s) which Prof Ward was asked to make about the results of the in vitro testing shown in the Poster. I address this topic at [309]-[318] below - see [315] in particular. I agree that the assumption(s) were unrealistic.
- The second point under this head concerned questions arising from [72] in his first report. The Claimants cited this passage:
“The Skilled Chemist would find it remarkable that it is possible to preserve the desired biological activity against the target, whilst modulating the PK and other properties, by making varied changes to the RHS substitution of the compound. That gives the Skilled Chemist a portion of the compound (the RHS substitutions) that they can vary and seek to modify, while retaining a degree of confidence that the desired biological activity will be preserved.”
- The cross-examiner was attempting to obtain a concession that methyl/fluorine groups could be appended on the right-hand side of RD7 or RD54. However, Prof Ward pointed out that his observation was made in relation to a comparison between RD37, RD131 and RD162. Underpinning his answers was the notion that the Skilled Team would not go back to RD7 or RD54 from RD162. I did not see anything in this criticism.
- The further (and more substantial) criticisms made in the Claimants’ Annex B are dealt with in the context of Astellas’ three reasons for inventiveness, to which I can now turn.
- Prof Ward’s opinion that there was no motivation to carry forward the disclosure of the Poster was a consequence of his assumption that the Poster represented a completed drug discovery and development project “marking the end of the journey” in which the authors have “reached their goal of developing an AR inhibitor for prostate cancer”, namely RD162, as he made clear in his first report.
- As the Claimants submitted, the basis for this opinion was unclear and in cross-examination Prof Ward accepted there was no statement in the Poster to that effect. Indeed, Prof Ward volunteered that it “may be an ongoing project or it may be a complete project” and “I accept that there are two scenarios which could be either that the team is carrying on work and is doing further analogues, or that that is the endpoint and they are taking that compound forwards. I think both of those could be possible” (XX Ward T4/38711-38813).
- In their opening, Astellas sought to make the same point, based on the arrows:
‘The Poster could not be clearer that it is describing a body of work that went from inception to lead compound in that research programme. To emphasise the clear direction of travel, the Poster is adorned with two large swooshing arrows - a feature which Astellas contended indicated a clear no turning back approach.’
- This submission was undermined by both medicinal chemistry experts who were clear that no scientific conclusions could be drawn from the arrows.
- Prof Clarke did not consider that RD162 was the ‘endpoint’ of the development of the RD series of compounds based on the disclosure of the Poster and he said there is no reason to believe that a decision has been taken to take RD162 forward into clinical trials, not least because it had not been tested for activity in vivo (XX Clarke T3/25814-2599). Ultimately Prof Ward accepted that the Skilled Team would not know that it was the end of the journey for the authors, not least because they have not tested RD162 in an in vivo efficacy model or using luciferase (XX Ward T4/47610-47920).
- In the course of his cross-examination, it emerged that a further aspect of Prof Ward’s reasoning was that the Skilled Medicinal Chemist would understand from the Poster that the cyclobutyl had been ‘actively’ selected and was therefore the preferred group at position X. On that basis, modifying RD162 to introduce a dimethyl substituent would represent a ‘backward’ step.
- The Claimants submitted this was a bizarre position for Prof Ward to adopt given that he did not know why it was the preferred group. Given the paucity of data presented he eventually accepted it was difficult to draw any conclusions as to the rationale behind the compound selected:
‘A. I guess it is a little bit challenging to know how rational and driven the process was as we only have a very snapshot of molecules presented. - XX Ward T4/38610-12.’
- Furthermore, Prof Ward’s view that the Skilled Medicinal Chemist would not be motivated to take the disclosure of the Poster forward was inconsistent with his description of drug discovery and development as set out in his own publications. His review entitled “Hit-to-Lead Medicinal Chemistry” (co-authored with Paul Beswick), identified (at section 2.1) several sources of ‘hit’ compounds including “molecules already known to bind to the target, for example, from competitor patent”. So, as he had to accept, the Skilled Team could use another team’s ‘lead’ compound as a ‘hit’ compound on which to build their own drug development programme (XX Ward T3/35917-36018). He was unable to explain why he had not considered this CGK approach to identifying lead compounds in his analysis on inventive step (XX Ward T4/45714-45811).
- The Claimants characterised this as an important omission, bearing in mind that Prof Ward also accepted that if the Skilled Team had identified RD162 as a starting point, they would want to make analogues of RD162 and compare the data from pairwise analogues in the manner described in the book chapter he co-authored with Stephen Connolly (XX Ward T4/45812-46323).
- The Claimants’ final point under this heading was that Prof Ward’s analysis on inventive step failed to take any account of the fact that the Skilled Team would be motivated to develop a novel compound (XX Ward T4/43724-44013). In Prof Ward’s review, the need to identify a novel candidate compound was described in Table 1 in his review as ‘essential’.
- The key issue however concerns what degree of novelty the Skilled Team would aim for. The point which was put (repeatedly) to Prof Ward was the Skilled Team would want to take forward a novel compound. It is true, as the Claimants’ submitted, that Prof Ward sought to avoid accepting this proposition when applied in the context of the Poster and RD162 and this was one of the Claimants’ major criticisms of Prof Ward, since the cross examination continued for some time (XX Ward T4/43313 onwards).
- Eventually, Prof Ward did accept that proposition (see T4/43824), but it was clear that he had not considered this in his reports. His evidence down to T4/45522 was very grudging in nature. For example:
i) Having said in his first report that RD162 was the end of the project, he seemed unwilling to take RD162 as a starting point for any further work by the Skilled Team ‘So if you are telling me I have to start with RD162….
ii) On that basis, he said he would seek to characterise RD162 to get a wider understanding of its potential, but immediately switched to making a back-up compound to RD162 ‘which would be a structurally distinct molecule’.
iii) It was then put that the first thing the Skilled Team would do would be to make analogues of RD162 to try and build up a SAR of the product, to which his response was that his eye was drawn to the right-hand side of RD162 as the place to make modifications.
iv) When the possibility of testing the dimethyl substituent was put, Prof Ward said he did not think that was an obvious thing to do ‘because there has been a selection towards the cyclobutyl’.
- In this whole passage of cross-examination, Prof Ward seemed to me to be bending over backwards to avoid accepting (either at all or without qualification) some fairly straightforward propositions, including some which he had previously written about in textbooks. He was also bending over backwards to avoid considering any changes at position X.
- On the basis of this passage of cross-examination, the Claimants submitted ‘Plainly, the desire for a novel candidate compound would itself be motivation for creating close analogues of the promising candidate disclosed in the Poster, namely RD162. This straight-forward proposition was put to Prof Ward who simply refused to engage, apparently unable (or unwilling) to understand the question being put to him.’
- Overall, in my view, there is no support for the ‘no motivation’ contention and I do not consider that Prof Ward’s point represented the position or views of the Skilled Team. Furthermore, there was a degree of exaggeration in Prof Ward’s suggestion that there had been an active selection of the cyclobutyl at position X. It is clear that the authors of the Poster had made the change from dimethyl to cyclobutyl and retained cyclobutyl. It would be fair to conclude there does appear to have been a preference for cyclobutyl, but there was no pairwise comparison nor any other data directed to this. On this point, Prof Ward was reading too much into the Poster, in my judgment.
- In his first report, Prof Ward said (at [139]) that “The Skilled Chemist would know that there are known metabolic liabilities associated with terminal methyl groups where oxidative metabolism can typically occur, and as such a terminal dimethyl substitution would typically be viewed as less metabolically stable than its cyclobutyl analogue” (emphasis added). In his second report he repeated this contention under his section on Inventive Step.
- I agree that it was clear that Prof Ward was putting this forward as a reason why the Skilled Team would not seek to make and test an alternative compound with the gem-dimethyl substitution. It was advanced as part of Prof Ward’s positive evidence in support of non-obviousness.
- In his oral evidence Prof Ward, faced with numerous contrary statements, including examples of dimethyl groups featuring in a number of commercial products, he sought to recast this evidence. He first said that he was not saying this was a reason not to go forward with the dimethyl just that if you see metabolism at this group cyclisation might be a solution.
‘Q. You see that. Did you consider it appropriate to draw to the court’s attention that a number of commercial products have gem-dimethyl groups when giving evidence that you give in paragraph 139; do you not feel that would be a more balanced picture?
A. I think we covered this ground yesterday as well. So what I am not saying is you should not incorporate a gem-dimethyl group. I am not saying that the gem-dimethyl group is one of those serious structural alerts that chemists are averse to, or actively avoid. What I am saying is if you have metabolism, if you see metabolism at the methyl of the gem-dimethyl group, then cyclisation would be an effective strategy to overcome that metabolism, if that is happening. What I was doing was trying to use my judgment of saying well why did I think there had been an active selection of the cyclobutyl. So I am not trying to advocate that the gem-dimethyl is not a group you can see in drugs or you can have. What I was trying to do was think "Well, why might it have been preferred?"’ - XX Ward T4/42013-4217 (my emphasis)
- As the Claimants submitted, Prof Ward then speculated that a possible explanation for why they went with the cyclobutyl is that they encountered stability problems with the gem-dimethyl. However, I agree that there was simply nothing in the PKDM data presented to support this. It was speculation. Moreover, it was not what he had written in his expert report (XX Ward T4/42523-43219). However, the Claimants submitted that Prof Ward had abandoned the metabolism of methyl substitutions as a factor in support of inventive step (XX Ward T4/4304-15), in the light of his answers under cross-examination.
- The Claimants made a number of submissions on this issue:
i) First, the gem-dimethyl substitution of RD162’ cannot meaningfully be described as a ‘terminal’ methyl group and there was no basis for suggesting that any concerns associated with terminal methyl groups also applied to gem-dimethyl groups (XX Ward T4/41810-41921).
ii) Second, Prof Ward was unable to point to any CGK materials in support of his view that there were known metabolic liabilities associated with gem-dimethyl groups (XX Ward T4/41922-4206; 42824-4296).
iii) Third, Prof Ward failed to mention that a number of commercial products have gem-dimethyl groups (XX Ward T4/4207-42212) or that he himself had written about a chemical series in which the preferred compounds all had gem-dimethyl groups with no mention of any concerns that they would give rise to metabolic liabilities (XX Ward T4/4228-42310) or that gem-dimethyl groups were widely used in medicinal chemistry without reference to metabolic concerns (XX Ward T4/42311-42419).
iv) Fourth, nilutamide a CGK commercial product in this area (Statement of Agreed CGK) has a gem-dimethyl substitution. Although nilutamide is metabolised by oxidation of the methyl groups the half-life of nilutamide was very long (56 hours) (XX Westwell T2/16521-16623; XX Ward T4/42420-42522).
- In my view, Prof Ward’s point had little or no substance. Furthermore, he raised it in a misleading way.
- The further point made in Prof Ward’s first report was that he said the Skilled Medicinal Chemist would consider that the “bulkier cyclobutyl substituent potentially filling better in such a pocket (at least relative to a gem-dimethyl substituent)” (Ward 1, para 138).
- Prof Ward alluded to this point again in the passage of cross-examination I mentioned above. At T4/453, Prof Ward said that when he saw RD162’ he was surprised that they had gone ‘backwards on the cyclobutyl to the gem-dimethyl’. He said: ‘There is an active selection of the cyclobutyl; that seems to me very clear’. Later he justified that in this way [T4/456]:
when you have what
11 looks to be an active selection of a group, and you have
12 references out to papers giving you a little bit more
13 information around homology modelling and structural work and
14 so forth, and papers which talk about various pockets you
15 might bind in, so you have work which seems to support that
16 active selection -- and again it is just hypothesising -- it
17 then seemed surprising that you can change that group back to
18 an earlier one and that then is a molecule which is going
19 forwards. I was surprised.
- At [153] above, I set out a summary of what the Skilled Team would derive from the papers referenced in the Poster, which was based on material set out by Prof Ward in his first report.
- It is clear, however, that the Poster makes no reference to a hydrophobic pocket of particular size, shape or dimensions. The Marhefka paper is referenced in a footnote. On p7, Marhefka reports that “One of the methyl groups on the hydantoin ring binds in the hydrophobic pocket below the 17 carbon of TES, and the other occupies the space of the 16 carbon of TES.” On the basis of this, Prof Westwell’s view was that the fact that only one of the methyl groups of nilutamide binds in the hydrophobic pocket would suggest it is something of a simplification to associate the bulk of the substituent with a ‘better’ fit into that pocket (Westwell 2, para 2.13). Prof Ward was unable to identify what (if any) aspects of Prof Westwell’s evidence on this point he actually disagreed with (XX Ward T4/4696-47418).
- Ultimately, Prof Ward conceded that this was no more than his suggestion as to why the cyclobutyl might have been chosen by the authors (XX Ward T4/4815-15). In other words, this was just a hypothesis. Ultimately it was clear that if the Skilled Medicinal Chemist identified this hypothesis, s/he would test at least the dimethyl variant against the cyclobutyl variant to see if this hypothesis was correct.
- On a side issue, I should briefly mention some diagrams which were put to Prof Westwell in cross-examination which purported to illustrate the size of the hydrophobic pocket. However, Prof Westwell was dismissive of these ‘cartoons’ and said they were a crude, two-dimensional schematic (XX Westwell T2/1322-10). Furthermore, there was no evidence that these diagrams bore any likeness to the actual shape of the hydrophobic pocket (XX Westwell T2/13020-13210).
- Overall, the position was that there was no information upon which to reason that the cyclobutyl was a better fit, other than the fact that the cyclobutyl had been adopted in RD37 and retained.
- Having considered all three of these points made by Prof Ward and the cross-examination on them, I find it difficult to avoid the conclusion that Prof Ward was, at times, engaged in a process of trying to find or generate arguments against obviousness.
- However, the mere fact that Prof Ward was so engaged does not mean the Claimants’ arguments succeed.
- The next part of Astellas’ case I must consider involves their attacks on the Claimants’ primary evidence and their contentions that the Claimants’ case changed as the trial progressed. This requires me to focus in more detail on the Claimants’ primary evidence of obviousness, as set out in the evidence of Prof Westwell.
- As far as I could detect, no challenge was made to the technical reasoning which Prof Westwell put forward in support of obviousness over the Poster. Instead, Astellas accused Prof Westwell of approaching the Pozzoli analysis incorrectly. They drew attention to 9.46-9.47 (and 10.28-10.29, which I consider later) of his first report and an answer he gave in cross-examination.
- The relevant context for the Poster was as follows:
i) At the start of his section on ‘Obviousness in light of [the Poster]’, in [9.42] Prof Westwell reminded himself (1) of the need to avoid hindsight, (2) to proceed without knowledge of the Patent and (3) to put out of his mind the knowledge he had from having read the Patent and the knowledge he acquired after the relevant date in his own research relating to enzalutamide. He then, in [9.43] set out an accurate statement of the Pozzoli approach.
ii) In [9.46], he identified correctly the difference between the Poster and claim 1. Then in [9.47] he stated:
‘For the following reasons, I do not believe that it would require any degree of invention for the skilled medicinal chemist who had read [the Poster] to make a modification to RD162 so as to make a compound where the cyclobutyl substituent was replaced by a dimethyl substituent.’
iii) His reasons were set out in [9.48]-[9.56] and I have summarised or set out those paragraphs above.
- Although I did not understand that the Claimants abandoned their primary evidential ‘immediately obvious’ case, it is true, as Astellas pointed out, that the Claimants did seek to develop a supplementary case: that it was ‘obvious to do a SAR’. It is possible that the Claimants had already anticipated the need to develop this prior to Prof Westwell’s cross-examination, but it is tolerably clear that the challenge made in his cross-examination cemented that perception.
- Although Astellas, through Prof Ward, ran a case that the Skilled Team would simply take RD162 forward, another equally obvious route is that Skilled Team would conduct some analysis (i.e. their own SAR starting from RD162), not least to develop their own molecule. What was in dispute was what would then happen.
- Although, as I have indicated, Prof Ward suggested that the Skilled Team would investigate modifications to the right-hand ring (the implication being only there), the Claimants suggested that Prof Westwell had agreed with this. They relied on this passage from one of his answers to a question I put to him:
“..it seems that swapping groups out of that right-hand aryl ring, largely, based on the limited evidence we have here, you retain activity. So long as you have that rigid phenyl group and you decorate it with different substituents, albeit a limited group, you are largely retaining the activity as an antagonist in that hormone refractory model” [T2/2018-16].
- In context, Prof Westwell was clearly talking about what appeared in the Poster. Later in the same answer, he did say this, but again, in my view, he was referring to what is shown in the Poster:
‘I think in the common general knowledge, or our respective reports, we both noticed that there seems to be a tolerance to a variety of substituents on the right-hand ring, that that is possibly more unusual.’
- So in their closing, Astellas tried to paint the picture that the Skilled Team would take one of two routes:
i) To continue to develop RD162 to pre-clinical and clinical testing and to make any further alterations along the way as and when problems arose;
ii) Alternatively, if the Skilled Team wants to make their own compounds based on the teaching of the Poster, then the obvious approach was to take the work that has been done and disclosed in optimising RD162 and then work on further optimisation of new compounds based on RD162 (probably focussing on the right-hand side of the molecule). Astellas’ point was that the Court cannot know what the outcome of taking this approach would be: such research is not pre-determined to an end result.
- In his cross-examination, it was put to Prof Westwell that when he had discussed the steps he had said were obvious over the Slides, he had not mentioned that the Skilled Team would do a SAR. His responses were that (1) SARs were discussed in the CGK section and (2) a medicinal chemist would do SAR - that is the nature of medicinal chemistry, an answer which appears in this important passage of the transcript at [T2/158-160]:
‘Q. …. I was
5 putting to you that, in both your first report and your reply
6 report, when you came to look at what is obvious to do in
7 light of the slides, and you have a heading "Obvious steps to
8 take in light of the slides", you did not talk about doing a
9 SAR, you did not set out what the goal might be, you did not
10 set out what might be tested, the order of testing, why one
11 might change just the gem-dimethyl and cyclobutyl at
12 position [X]. You did not discuss what else might be done. You
13 did nothing. What you said was "Well, it is an obvious
14 change". I am putting to you that is all you thought about at
15 the time, you did not think of doing any SAR, large, small,
16 medium or otherwise?
17 A. Yes, but a medicinal chemist would do SAR. Whether it is
18 large or small, that is what they would do. They would be
19 interested in further exploration around the substituent
20 groups which would include the hydrophobic region as well as
21 the aryl. That is the nature of medicinal chemistry.
22 Q. As we discussed, one way that they would do that is either
23 because they are taking forward RD162 and they hit problems,
24 or they may have a target, given to them by the biologist, or
25 they may have another set of targets that they wish to do, a
Page 159:
2 bit like you did, in their research. Of course, you have not
3 discussed any of those in your evidence, have you?
4 A. No, because I was asked to assess obviousness in terms of --
5 you could certainly do exploratory SAR and make more wholesale
6 changes but in terms of this hydrophobic region, it is
7 reasonable, on PK grounds, any premises like LogP, that these
8 changes in the hydrophobic region having or taking away a
9 single carbon atom would not materially affect those
10 properties. So, yes, I did not discuss SAR building on 162 in
11 the expert reports because that could be done but that might
12 involve more substantial changes to the substituents.
13 Q. I think you said you did not because you were asked to assess
14 obviousness in terms of -- I am assuming what you were going
15 to say is in terms of the difference and assessing the
16 difference between a gem-dimethyl and a cyclobutyl?
17 A. Mmm-hmm.
18 Q. And with that change in mind, you were then saying well, is
19 that going to have an effect or not or make any obvious
20 changes? That is what you were doing really, is it not?
21 A. Yes, assessing the obviousness of that change in light of the
22 material presented in the public conference, yes.
23 Q. That is right. So when you say the obviousness of that
24 change, what you were thinking was "If I am asked about the
25 change between cyclobutyl and gem-dimethyl, do I think that
Page 160:
2 change is an obvious one to make in light of what I have
3 here?"
4 A. Yes.’
- Later, when asked about his evidence as to what was obvious over the Poster, Prof Westwell confirmed that he had approached that issue in the same way as he had on the Slides and came to the same conclusion.
- Three important points emerge from that passage:
i) First, that when challenged, Prof Westwell did not say that his reasoning was part of a SAR.
ii) Second, that Prof Westwell regarded the obvious steps he had talked about in his reports as distinct from a SAR.
iii) Third, that he was asked to assess obviousness in terms of the difference between a gem-dimethyl and a cyclobutyl. However, I keep in mind that when Prof Westwell said this, he might well have been referring to Pozzoli step 3 (identify the differences). As I have already mentioned, he set out the Pozzoli approach in [9.42] of his first report and there are clear signs in his written evidence that he was endeavouring to follow that approach.
- Astellas submitted a fourth important point also emerged from the final question and answer: that in that answer Prof Westwell effectively admitted that he had the target (i.e. RD162’ i.e. with the dimethyl group) in mind when concluding the step from RD162 was obvious. However, I do not consider that was clearly established. There is a subtle difference between having the target clearly in mind and considering whether it was obvious to make a change at position X from the cyclobutyl group to a dimethyl group. The basis of Prof Westwell’s penultimate answer was ‘in light of the material presented in the public conference’ (which would not have included RD162’). That was echoed in the final question.
- Astellas nonetheless submitted that it was clear from his written reports that Prof Westwell’s analysis was infected with hindsight. The basis for this suggestion was the following. One starts with the agreed CGK that at the start of a drug discovery project the Skilled Medicinal Chemist and the Skilled Cancer Biologist would agree a TPP (see [97] above). In cross-examination, Prof Westwell agreed that what the Skilled Medicinal Chemist would do would be conditioned on the TPP to be discussed with the Skilled Cancer Biologist, although there may be considerable variation in the amount of knowledge about the starting molecule. The SAR would progress to identify a front runner molecule, whereupon modifications are likely to become more focussed on improving that front runner, although you are also building up a model of the chemical structure requirements for binding to the desired target. The SAR progresses to identification of a lead molecule which has a range of properties across the board, giving the Skilled Team confidence to progress to more advanced testing. Problems may be encountered leading to alteration, substitutions and further research directed at trying to overcome them. It is also prudent to develop back-up compounds, designed to mitigate a potential issue with the lead compound.
- Overall, Prof Westwell agreed that a SAR is quite a wide umbrella term since what needs to be done in the SAR depends on how much is known at the outset, what the goal is, how much testing has been done and so on. He also agreed that that was very much the work of the Skilled Medicinal Chemist ‘to have that understanding of the issues that you have just outlined, and to design a programme of optimisation of structural activity study to hopefully overcome any challenges..’
- Against that background, Astellas point out that, when it comes to the evidence on obviousness, there is no mention of a TPP either by Prof Westwell or Prof Hickson, even though it is supposed to be agreed between the Skilled Medicinal Chemist and the skilled cancer biologist. Furthermore, in his written evidence Prof Westwell did not expressly mention conducting a SAR exercise - hence the cross-examination from which I quoted the key extract above. See in particular T2/15817-15912.
- So, the problem with Prof Westwell’s evidence on obviousness is the absence of a proper description of the context in which his Skilled Team formed the view that it was ‘immediately obvious’ (from the Poster) that the two RD162 analogues he depicted at [9.51] would be likely to have therapeutic potential that was similar to that of RD162.
- It could be said that the Professor was presented with an unusual situation, in that the Poster presented a molecule RD162 with all the attributes of a lead molecule, so there was no need for a TPP or a SAR to be conducted. Yet Prof Westwell never said as much. Equally, Prof Westwell did not say that the Skilled Team would embark on an exercise of seeking to characterise RD162 by making analogues (this being a point which was put forcefully to Prof Ward). He did explain why the Skilled Medicinal Chemist would have been either reluctant or uncertain about making modifications to the other parts of the molecule, apparently leaving only position X to be considered.
- Furthermore, the absence of a SAR or other context could be said to be emphasised by the fact that in [9.53]-[9.56] he considered whether changes to other parts of RD162 would result in molecules expected to have similar therapeutic potential, but apparently only because he was asked to do so i.e. that consideration did not arise because of a particular scenario or context that his Skilled Team was considering.
- At this point it is necessary to consider the sequence in which Prof Westwell was introduced to the various documents in the case. It is clear that the sequence was: consideration of the CGK of the Skilled Medicinal Chemist; what the Skilled Medicinal Chemist would understand from the Slides; then he was shown the Patent and asked to explain what the Skilled Medicinal Chemist would understand from its contents. He explained that many months later he was provided with a copy of the Poster and was asked to describe what the Skilled Medicinal Chemist would understand from its contents, having been told to put out of his mind what he had previously discussed with the solicitors.
- Prof Westwell acknowledged that he was familiar with the compound now known as enzalutamide from his own work in the field from 2014-2020, and that he was told to put out of his mind his personal knowledge acquired after March 2006 and he stated he was satisfied that his knowledge of enzalutamide had not affected his opinions.
- Despite the sequence, it is curious that Prof Westwell’s report was structured so that he presented his views on obviousness over the Poster first (with the ‘immediately obvious’ wording). When it came to obviousness over the Slides, he did not use that expression and his reasoning was tied much more closely to material in the Slides, as I discuss below.
- The ‘obvious to do a SAR case’ was enthusiastically developed in the cross-examination of Prof Ward with a number of approaches being suggested which I will now summarise. Some of the suggestions put were bizarre: ‘they may just have had plenty of cyclobutyl in the fridge’.
- It was put to Prof Ward that a Skilled Team will not want to go with one of their competitors’ products, they would want to create their own novel products. This was based on the article he had written mentioned above setting out the process of drug discovery in which he had indicated that novelty was essential for hit, lead and candidate compounds. He agreed that if one had done a high throughput screen and obtained two lead series or two hit series and all other things were equal, a team would “naturally go for the one that had more novel structures in it than not” [T3/3628-24]. He qualified what he had written on the basis that it is ‘a holistic judgment’ of all the properties. The Skilled Team is trying to understand what is the potential for turning the hit series into a candidate, it maybe that it is not novel but the team can see a very clear strategy to make it novel in terms of where they are going on their optimisation journey [T3/3632-3642].
- It was put to Prof Ward that the patent department might ask the chemist to look for the next most suitable analogue, because the chosen candidate had been patented by someone else. Prof Ward’s initial reaction was “that is not a situation I have ever encountered”. As Astellas submitted, that is not a promising start for this attack, particularly since Prof Ward has considerable “real world” industry experience at Cerebrus and then GSK. Prof Westwell accepted that Prof Ward’s experience was far more industry focussed than his own [T2/9820-23]. Astellas submitted that if anyone would have had instructions from a patent department to look for the next most suitable analogue for patent protection reasons, it would have been Prof Ward. Yet, he had never encountered this particular scenario.
- Prof Ward explained that if the instructions had come from the patent department to make something new and protectable, the Skilled Team would be more likely to focus on something structurally diverse - in order to achieve something protectable [T4/4519-4524].
- This led to the proposition becoming more extreme. He was then asked to assume that the patent department came and said “We do not care how close it is; it just has to be different to 162, as close as you like…”. Even on this basis, Prof Ward said he would not know what underlying patent application or applications had been filed by the authors, which would lead him to go further away structurally. At [T4/4525-4539] he explained:
‘A. My concern would still be that either there has been a series of close analogues that have been made and discarded, and I would want to try and understand why those had been discarded. I would still be concerned about, notwithstanding the patent or (unclear), from a novelty perspective. I would be aware that the patents potentially have not appeared yet so we do not know what molecules, what compounds, have already been claimed. So, all of those things would encourage me to be further away structurally.’
- This line of questioning based on instructions from the patent department was, in my view, plainly hindsight driven. If patenting considerations are to be taken into account, the patent department would consider it was highly likely that a patent application had been filed covering the research disclosed in the Poster which would feature a familiar cascade of claims, often initially claiming a Markush structure but then increasing in specificity to a claim to the preferred molecule. That is precisely what featured in the PCT application which led to the Patent. In fact (as often occurs) the wider claims were abandoned and the Patent claims only the single compound RD162’, in this case because of the prior publication of the Poster, but the patent department would not have known this at the relevant date.
- Furthermore, the normal expectation of the Skilled Team would be that the widest possible patent protection would be in the process of being sought and that would normally indicate that the development of a novel (and protectable) molecule from the starting point of RD162 would require some significant modifications. So, if one strips out hindsight, the quest for novelty would take the Skilled Team much further away from RD162 than the change from cyclobutyl to dimethyl.
- This was another route suggested in cross-examination of Prof Ward. It is the idea that a Skilled Team will look at the Poster, not proceed with RD162 or any optimisation of that, but will instead seek to undertake a SAR on RD7. Astellas submitted that the reason why the Claimants alight on RD7 is plain (it has a dimethyl) but also that there is no reason for a Skilled Team (without knowledge of the Patent) not only to suddenly start from RD7, but then to adorn it with the right-hand side substituents of the compound from where they had chosen not to start (RD162), and not change anything else. They contended this is another hindsight analysis.
- Astellas contended that the Claimants’ problem with this line of argument is that there is no reason to want to progress RD7 and no direction as to what would be done if it was progressed. I agree, for the following reasons which Astellas developed in their closing submissions.
- On the face of the Poster, RD7 has worse activity than RD37 both in the in vivo assay on the right-side of the Poster and in the PSA level assay at the bottom. The clear evidence is that a Skilled Team would wish to progress the best compound. Prof Ward explained that “from my experience, you would have a team sitting together looking through the data. You just have so many assays. There will be a compound which is preferred from that set, and you would take that compound forwards.” [T4/3808-11].
- From the data in the Poster, that compound was RD37 which was optimised through RD131, RD161 to RD162. But the potential for further optimisation on the right-hand side of the molecule is clear in order further to improve PK and drug like properties. Why go back to a discarded compound with ostensibly less activity?
- Apparently in an attempt to try to avoid this being branded as hindsight-driven, the case was put on the assumption that there is no material difference (although sometimes it was put as no difference: [T4/39712-39824]) between RD37 and RD7 (and sometimes between all RD compounds from RD6) due to the lack of any statistical significance in the absence of error bars.
- There is no dispute that the lack of error bars means that the data have to be interpreted with some caution. The evidence on these is as follows:
i) Prof Clarke pointed out that the trend in the in vivo tumour assay in the Poster was showing RD37 as having greater effect but “I would be circumspect in over-interpreting that” [T3/24524-2469].
ii) As regards the PSA data in the Poster, Prof Clarke thought that RD37 was more potent than RD7, particularly at the highest concentration, and would want to take forward the compound with the best activity at the highest concentration [T3/25919-2659].
iii) Prof Hickson viewed the graphs alone “without following that story” and simply reviewing the graphs “in isolation”. On that basis, the lack of error bars on the in vivo assay in the Poster meant that in respect of this assay he could say no more than RD37 appears to be “modestly more active than RD7” [T2/6915] and he “assumed that if you did the PK with those, they would also be roughly equivalent” [T2/709-12].
iv) However, Prof Hickson was better able to interpret the PSA level graph at the base of the Poster. In §7.18-7.19 of Hickson 1 he had not had difficulty with error bars or such like, and commented on the fact that RD131 appears to have the best activity at the highest concentration and that the others were maybe equivalent depending on the concentration compared. In cross-examination he confirmed that he agreed with Prof Clarke about the importance of the best activity at the highest concentration in a prostate cancer drug [T2/8817-9115].
- Overall, the experts agreed that whilst RD37 looks slightly ahead in the tumour volume study, it is difficult to draw conclusions, but RD37 and RD131 are both better than RD7 in the HR prostate cancer PSA level assay - something that the biologists agreed would be of real interest.
- However, the Claimants never explained why a Poster at a conference would be expected to have error bars on it or information to enable the reader to make statistically sound conclusions. The Poster was just giving a gist of what has gone on. The gist here is that RD37 was the best antagonist to take forward to in vivo PK testing, but its half-life and steady state then proved to be poor. That then tells the story of the work done on the right-hand side of the molecule to improve the drug like properties.
- Astellas submitted that the Claimants’ approach was to focus on that fact that the Poster does not permit a statistical conclusion to be drawn, and then seek to conclude from this that no conclusions can be drawn. The one does not follow from the other and it ceases to look at the teaching of the Poster as a whole.
- Although no statistical analysis has been done, as Astellas submitted, the clear take home message of the Poster is that RD37 had better activity than RD7 which was why it was RD37 that went through to in vivo PK testing and it was why it is the scaffold structure of RD37 which was then shown being changed with the different right-hand side substituents.
- Notwithstanding the above, the case was put to Prof Ward on an assumption that a Skilled Medicinal Chemist reading the Poster would think all of the compounds after RD6 were essentially the same or the same. The assumption was something of a moving feast:
- At T4/395 - it was that the authors of the Poster had formed the view that a cyclobutyl or dimethyl or cyclopentyl were all equally suitable.
- At T4/397 - it was that RD37, RD54 and RD7 all perform equally well and have materially the same IC50s.
- At T4/398 - it was that RD7, RD37 and RD54 are all equally active.
- At T4/400 - they are all much of a muchness.
- At T4/404 – it was that they are all, broadly speaking, the same, metabolised in a similar way, you have to choose one.
- At T4/415 – it was that there is no difference between these compounds in the antagonist assay.
- At T4/416 - it was that the data points of RD6-RD162 are all equivalent in the HR prostate cancer cells.
- At T4/449 - it was that there is no difference between RD7 and RD37 for the purposes of this question.
- I agree that all of this was untethered to reality. A Skilled Team reading the Poster as a whole would understand that the message being conveyed is that the most active compounds were RD37 and RD131 and work had then been done to improve their “drug like” properties leading to RD162.
- If all the compounds are treated essentially equally or even equally as advanced in cross-examination, that creates a number of problems for the Claimants’ case:
i) First, if (as the assumption sometimes required) the molecules are thought all to be “similar”, then the Skilled Team would pick the best to take forward. There is no basis at all for saying that this would be RD7 (Ward [T4/40410-13]).
ii) Second, if (as the assumption sometimes required) the molecules are all the same - then there is no way of choosing between them. On that basis, further testing would have to be done in order to select a front-runner - otherwise no active selection is being undertaken at all - it is essentially random, something Prof Ward described as a “very unrealistic proposition” [T4/39616-21]. If in fact the compounds cannot be differentiated on the data in the Poster, then the Skilled Team would do further in vitro tests (including solubility and permeability testing) in order to differentiate them (Ward [T4/4012-40324]. There is no evidence as to what might then happen and where that might then lead.
- If the data in the Poster are to be ignored (or treated as meaning that all the RD compounds are basically the same), Astellas submitted that the Court has no idea which might be taken forward and what changes might be made. On this basis it might be RD6 but who knows what might be done with the azide group. It could be RD54 with different substituents building on the cyano group. It could be RD7 but it might not be and who knows what substitutions a Skilled Team might make.
- Even if RD7 was selected, and was found (contrary to the indications on the Poster) to be the most active and so worthwhile pursuing, there is no reason at all to keep the entire right-hand side of RD162 identical, considering that the whole point of going to RD7 in the first place is because the Skilled Team did not want to proceed with RD162. As Astellas submitted, the Claimants’ case was circular. They posit a Skilled Team that does not want to progress RD162, who go to RD7 but then seek to make a very close analogue of RD162. Astellas submitted this makes no sense.
- Finally, it was suggested that maybe RD162 would be progressed but RD162′ might be alighted upon as a back-up compound. Prof Ward explained this would be “super risky” [T4/46510-46617]. For similar reasons as explained above, Astellas submitted that nobody makes back-up compounds (the insurance for a rainy day with the main compound) with a goal of them being structurally similar to the candidate compound. The idea is to have something structurally diverse.
- Before I reach any conclusions, I must finally consider three points on secondary evidence.
- First, Astellas rely on evidence of what Professor Westwell himself did when faced with enzalutamide after the application date and their contention that there was no suggestion that the approach to SAR dramatically changed from 2006 to 2016, which was the date when Prof Westwell worked on enzalutamide after it was launched in 2012.
- Astellas submitted that in none of his papers did he even once change the substituent at position X on the central ring. In one of his papers, the team of which he was a member sought to improve the antiproliferative properties of enzalutamide and looked at exploring the right-hand and left-hand rings, adding a 3,5-bis-triflouroumethyl group in ring B, a bulkier 3,5-tert- butyl was envisaged as well as adding further alternative substituents in ring A. Prof Westwell depicted the approach in fig. 1 of [XX-AW/8/103] set out below:
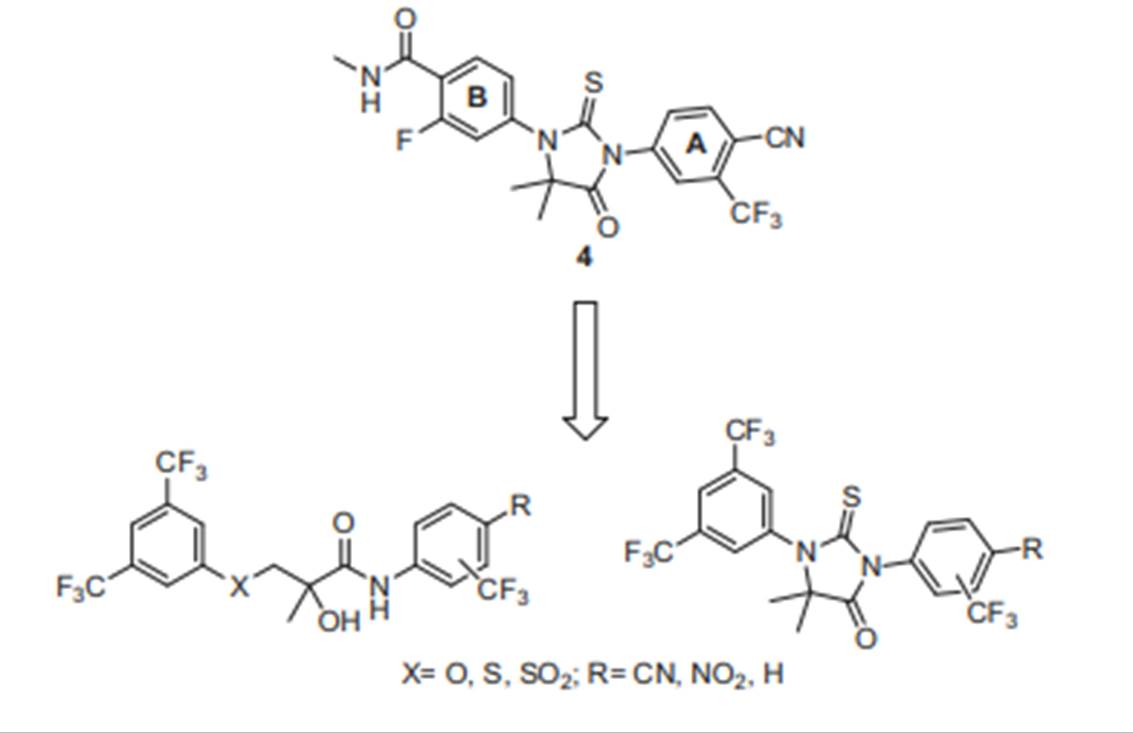
- The compound at the top is enzalutamide and bottom right are his proposed changes to enzalutamide (bottom left is bicalutamide). No changes are made to position X on the central ring - the focus is on rings A and B. At no point in any of Prof Westwell’s papers do we see any change made to position X on the central ring. Astellas invite the conclusion that this entirely corroborates the opinion of Prof Ward, who would have optimised the right-hand ring. They suggest it is the opposite of the secondary evidence in Monsanto v Merck.
- I do not propose to place any weight on this point because the context(s) for the later work done by Prof Westwell were different.
- Second, in their opening, the Claimants sought to rely on a separate point which concerned how in fact the inventors came up with RD162’. I would not have referred to this topic at all (because it is completely irrelevant) but for the fact that Astellas sought to rely on some statements made in emails between some of the inventors as secondary evidence that the Skilled Team would not predict that the change to a dimethyl would not make a material difference.
- The email from Dr Jung on 9 February 2006 explains that testing was being done on RD162′ and RD162′′ and he commented “let’s hope they both will have very similar activity and PK profiles to those of RD162”. In the earlier part of the same email chain of 6 February 2006, he had added a postscript: “PS. let’s hope RD162′ is as active as RD162!”
- Astellas point out that the inventors were steeped in knowledge about the RD compounds but even they were uncertain as the effects of the changes featured in RD162’ and RD162”.
- In the absence of any evidence from those involved, it is necessary to consider the relevant context in which these comments were made. The Poster (and the Slides - in so far as these actions are concerned) were made available to the public at the Prostate Cancer Foundation Scientific Retreat in Arizona that took place between 29 September to 1 October 2005. The filing date of the Patent was 29 March 2006. The emails were sent in early February 2006. By that time, it is a reasonable inference that the inventors had considerably more data about RD162 than had been published. When Chris Tran said ‘PS. I am curious as to why we are having a modified version of RD162. RD162 is really good already’, it is likely that the inventors already had full data on RD162 and had probably selected it as the molecule to take forward, but for this last-minute hiccup.
- I consider this point is double-edged. On the one hand, one might say that the inventors chose to investigate RD162’ and RD162” precisely because they considered the changes which those molecules featured would not make any material difference to the activity and PK profile of RD162. Of course, that could not be guaranteed so the new molecules had to be made and tested.
- On the other hand, Astellas may have a point here, but I am not persuaded I should take it into account. I am not convinced this scenario can be treated as a proper analogy to the scenario faced by the Skilled Team having read the Poster.
- Third, Astellas developed some points in cross-examination based on particular compounds in the Tiers in the Patent. Various changes at position X away from cyclobutyl took certain compounds down into Tier 4 (no better than bicalutamide). I am not sure that Astellas actually sought to rely on these examples as secondary evidence that small changes can have a big impact. The Skilled Team would readily accept they can, but it depends on the circumstances. Furthermore, these points confuse two separate things: The first is a reasonable prediction which the Skilled Medicinal Chemist makes based on certain data. The second concerns the result(s) of the testing of the prediction which may or may not turn out to be accurate. Just because the results do not conform to the prediction does not mean that the prediction was unreasonable or that it was not worth testing.
Conclusion on Obviousness over the Poster
- I am acutely conscious that there is a lot riding on cases of this type which leads to the legal teams and their expert witnesses going to great lengths to establish their own case and to damage the case being presented by the other side. This can lead to over-complication and, in my judgment, it has done so in this case.
- This is not entirely the fault of the parties. When an allegation of obviousness is made, the Court is obliged to adopt the viewpoint of the notional team of ordinary skill in the art, assess their common general knowledge and then decide the question of whether the step(s) from the prior art to the claim would have been obvious to that team, acting without hindsight. We are told that the notional Skilled Team should mirror teams in real-life, albeit with suitable adjustment to ensure the notional team is not inventive. The process is not automatically insulated from over-complication, but consideration of what would happen in real life can help to eliminate it.
- In this case, the over-complication has occurred because there was much theorising as to what the effects of certain mooted changes to RD162 would be. I have no doubt that real-life Skilled Teams would debate their theories of action - up to a point. However, in real-life teams, the theorising would quickly give way to action to test whether one or more of the theories had merit (assuming, of course, that pursuit of the theory was considered to have a reasonable expectation of success).
- This case involves a somewhat unusual situation, in that what was published, in the Poster was a molecule RD162 which showed good efficacy and good PK-DM properties. It was clear that the Skilled Team would regard RD162 as a good candidate to take forward into further testing. The motivation to consider other molecules came from competitive and patenting considerations, in the sense that it would be prudent for the Skilled Team to view RD162 as ‘belonging’ to the authors, it being a reasonable assumption on the part of the Skilled Team that the authors/their employers would have already applied for patent protection, and therefore the Skilled Team needed to develop their own molecule, novel and different from RD162.
- This case raises, in an acute form, the issue as to the extent to which competitive and patenting considerations should influence an obviousness analysis. As usual, the answer is provided by consideration of what real-life teams would do. Although some expert witnesses do have experience of patenting considerations, many do not, so the Court can be left to rely on its own experience.
- I can start with the case as originally presented in Prof Westwell’s first report. There are a number of problems with this case, including the following:
i) Prof Westwell does not explain the scenario in which the Skilled Team decides to take the step which he says is obvious. In particular he does not explain why the Skilled Team starts by considering position X, nor why the Skilled Team starts to consider changing the cyclobutyl group at that position.
ii) His answers in cross-examination indicate that he was not considering the situation where the Skilled Team had decided to do a SAR or characterise RD162 in order to find out information which would help to decide how to proceed. Instead, his answers indicated he was asked to consider the change from cyclobutyl to dimethyl and asked whether that change would have a material effect. Even if I assume in his favour that he was referring to Pozzoli step 3, the invitation put to him was leading.
iii) In this regard, there is no separate body of law which is applicable if the change required to get from the prior art to the claim is said to be ‘immaterial’ or ‘trivial’. In every case, there is only one question and it is the statutory question of ‘is it obvious?’. Whether a change is properly characterised as ‘immaterial’ or ‘trivial’ is highly fact dependent and there is a real danger (as has been pointed out in some of the authorities) that the question of immateriality is substituted for that of obviousness, even though a conclusion that a change is immaterial or trivial will usually be determinative of the question of obviousness. In general terms, I agree with Astellas’ submission that these issues of ‘immateriality’ or ‘triviality’ are not well suited to the field of medicinal chemistry, but it all depends on the context.
iv) The quest for a novel compound would have taken the Skilled Medicinal Chemist away from close analogues to RD162. The focus on close analogues was driven by the hindsight knowledge that the Patent in fact claims RD162’. Without that knowledge, on this primary case the Skilled Medicinal Chemist would not have been motivated to investigate close analogues of RD162. I have to consider this point further in the alternative ‘obvious to do a SAR’ case.
v) Overall, I am driven to the conclusion that Prof Westwell’s evidence that the change from cyclobutyl to dimethyl was ‘immediately obvious’ was tainted with hindsight.
- On that primary case, I did not find persuasive any of the three points which Astellas advanced in their positive case of inventiveness. In my view, Prof Ward was trying too hard to generate points in favour of Astellas’ case.
- I turn to consider the alternative case - it was obvious to do a SAR. Having read and considered the disclosure of the Poster with interest, I find the Skilled Team would have been motivated to do a SAR. I dismiss the idea that the Skilled Team would use any compound other than RD162 as their starting point. With RD162 as the starting point of the SAR, I also find the Skilled Team would have been motivated to develop a novel compound from that starting point. The real dispute lay in where that process would lead the Skilled Team.
- As I mentioned, Astellas submitted there were only two options for the Skilled Team: take RD162 forward or do a SAR in order to develop a structurally distinct molecule. The Skilled Team starting on a SAR based on RD162 would wish to characterise RD162 i.e. to develop their own (and better) understanding as to how various parts of the molecule contributed to its properties. As Prof Ward put it: ‘…. I would characterise it, and not just characterise it to reproduce the data that are here, [i.e. in the Poster] I would characterise it to get a wider understanding of its potential …’.
- This was the furthest that the evidence went on what would be done in the SAR. At best, it remained unclear whether the Skilled Medicinal Chemist would undertake a full SAR investigation (which would include making and testing RD162’) or whether s/he would accept some of the points indicated in the presentation in the Poster, and if so, which ones (which might result in RD162’ not being made or tested). As I mentioned at [290] above, Prof Westwell agreed that a SAR is quite a wide umbrella term since what needs to be done in the SAR depends on how much is known at the outset, what the goal is, how much testing has been done etc. For the SAR argument to succeed, it was necessary to spell out the goal and what testing would be planned, against the backdrop of RD162 as the starting point and the information in the Poster.
- This alternative case depended on making and testing RD162’ not as the or one of the target molecules having conducted the SAR, but almost as a byproduct in the early stages of characterising RD162. It is possible to envisage that, on other evidence, that process could have led to a finding of obviousness. The problem for the Claimants is that one normally expects a true case of obviousness to be set out clearly in the evidence in chief of the party making the allegation. This argument was not set out by Prof Westwell at all. The evidence given by Prof Ward came very close but I was not satisfied that the target was established. Once again, I find the intense focus on the change from the cyclobutyl to the dimethyl was tainted by hindsight - the knowledge that RD162’ was the target.
- In one sense, Prof Ward’s dogged resistance in cross-examination on the points I considered above might have favoured the Claimants’ case, but it cannot improve it largely because the cross-examination was itself driven by the focus on reaching the target. For the avoidance of doubt, I placed no weight on any of the secondary evidence.
- In the circumstances, I reject the allegation that claim 1 was obvious over the Poster.
- Save for one point, there was no real dispute as to what the Slides disclosed to the Skilled Team.
- The Skilled Team would understand from the Slides that the authors were investigating compounds for use in the treatment of HRPC. They would further understand from the Slides that it was important for such compounds to be AR antagonists and not to act as agonists in HRPC.
- It was common ground that the Skilled Team would consider the cross-referenced Chen paper on Slide 1. The material presented on Slides 2 and 3 regarding AR overexpression would be understood to be material taken from that paper.
- Slides 5-7 describe the general approach of the authors in seeking compounds that were stronger antagonists of AR than bicalutamide, but without showing agonist activity in cells that overexpressed AR.
- Slide 8 is a summary of “Design, Synthesis and SAR studies” that the authors have carried out. This was agreed to be a key slide.
- It explains that certain design tools were used to initially arrive at the compound shown as RU59063. This has high AR binding affinity but agonistic activity. Prof Westwell described the structure of RU59063 by reference to circled parts shown in the figure below:
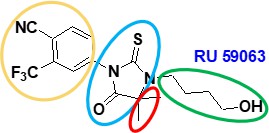
i) the yellow ring shows a 6-membered aromatic ring (phenyl) with two substituents: a cyano (CN); and a trifluoromethyl (CF3) group. The Skilled Medicinal Chemist would note that the same disubstituted phenyl ring is present in bicalutamide (see structure on Slide 5).
ii) the blue ring shows a heterocyclic five-membered ring with sulphur and oxygen substituents, known as a thiohydantoin.
iii) the green ring shows a hydroxybutyl (C4H8OH) group bound to one of the nitrogen atoms of the thiohydantoin ring.
iv) the red ring shows two methyl groups (a geminal dimethyl group) at the bottom of the structure bound to a carbon atom of the thiohydantoin ring.
- Slide 8 also shows a pharmacophore (shown below). As Astellas pointed out, a pharmacophore should not be confused with a Markush formula: instead, it provides an overview of the authors’ views of different interactions of a molecule within a binding site (in this case, the interactions of the molecules under study within the AR binding site). The targets for investigation are marked R, R1 and R2.
- The Skilled Medicinal Chemist would note the following about this figure:
i) The left-hand side (LHS) of the pharmacophore indicates that the cyano group (NC) at the 4 position of the LHS phenyl ring is important for hydrogen bond interaction and that the trifluoromethyl group (F3C) at the 3 position of the LHS phenyl ring is important for hydrophobic interaction. The LHS of the pharmacophore is structurally the same as the LHS substituted phenyl rings of both RU59063 and bicalutamide.
ii) The LHS phenyl ring (shaded yellow) and part of the middle thiohydantoin ring (shaded turquoise) are indicated as being important for binding affinity to the AR. This part of the thiohydantoin ring is structurally the same as the thiohydantoin ring of RU59063. Bicalutamide does not have a thiohydantoin ring.
iii) The left-hand side of the structure (in yellow and blue) are indicated as important to binding affinity to the AR and shown as fixed (i.e. no change).
iv) The remaining part of the middle thiohydantoin ring (shaded red) is indicated as being important for hydrophobic interactions with the AR. The geminal dimethyl group (red) is generalised from RU59063 by the use of two further placeholder groups, shown by R1 and R2.
v) The right-hand side (RHS) (shaded green) is another phenyl ring and is said to provide “antagonist activity”, with the phenyl ring also providing “rigidity”. The Skilled Medicinal Chemist would understand that rigidity is obtained because a phenyl ring is a flat and rigid group that can only rotate on its axis. This contrasts to RU59063 (which is said to have agonistic activity) which did not have rigidity in this position, as its flexible alkyl chain can take on lots of different configurations. The experts agreed that the Skilled Medicinal Chemist would therefore understand that the key development in this structure over RU59063 is the introduction of the “right” (green) phenyl group, with this moiety seemingly resulting in the molecule having the desired antagonistic activity without agonistic activity.
- Slide 9 shows the structure of RD37 and describes it as a potent antagonist in hormone sensitive LNCaP cells. Data is presented that shows RD37 reducing relative PSA levels in a dose dependent manner compared to bicalutamide and RU59063. RD37 exemplifies the pharmacophore of slide 8 in that:
i) In place of R1 and R2 at the C5 position of the middle thiohydantoin ring is a cyclobutyl (a four-membered carbon ring).
ii) In place of R at the 4 position of the RHS phenyl ring is a methyl group (CH3).
- Slides 10-15 present further data relating to RD37, setting out its benefits, including that it acts as an AR antagonist in HRPC cell lines without agonist activity (slides 10, 11), that it shows selectivity for AR (slide 12), that it has comparable binding affinity to bicalutamide (slide 13), and that it slows the growth of HRPC cell line in vivo compared to bicalutamide. Slide 15 provides some PK and PD data on RD37, showing that it has a short serum half-life and is cleared after around 6 hours.
- Slide 16 presents the structures of 2 derivatives of RD37: RD131 and RD162. It also presents PK data relating to these compounds and also bicalutamide. This is materially the same as the data that is shown in the Poster (see [173] above), the only difference is that the text below the chart on slide 16 makes it clear that the serum concentrations were measured after IV dosing, whereas this is not stated in the Poster. This slide shows that RD37 has PK problems, with high logP values (too lipophilic) and its PK after dosing (shown in the graph as the light blue line at the bottom) is poor (almost all gone after 15 hours). No steady state value can be given.
- The box on slide 16 indicates that the inventors had then tried a number of different compounds (the structures of RD131 and RD162 being shown) with RD162 having good IC50 values, reasonable LogP and good steady state concentration, when compared to bicalutamide. Of the three molecules disclosed on this slide, RD162 performs by far the best in PK after IV dosing, where it shows promising performance as compared to bicalutamide.
- Slide 17 shows how RD37, RD131 and RD162 perform in a cell-based assay mimicking HRPC, measuring relative PSA levels. RD37, RD131 and RD162 all show an antagonist-type dose response with increasing concentrations.
- Slide 18 states the following conclusions:
‘Cell-based screens can be used to identify anti-androgens with greater potency than bicalumatide [sic] while avoiding the undesirable agonism side-effect
SAR has defined a thiohydantoin imine derivative of the high affinity ligand RU59063 as an attractive lead
Greater potency can be achieved in the absence of greater binding affinity, presumably through inducing altered AR conformation
Further in vivo studies are in progress to define an optimal clinical candidate’
- The experts were agreed that the first three conclusions summarise the key points from the preceding slides (even though the reference to ‘imine’ would not be understood).
- Astellas suggested that the changes made to RU59063 might have assisted in improving the potency due to “inducing altered AR conformation” - in other words the changes made at R and R1 and R2 have potentially altered the conformation of the receptor leading to improved potency.
- Prof Westwell thought that the greatest contributor to this improved potency was the installation of the rigid aryl ring, but he accepted that the change to cyclobutyl “is part of that. It has a role in binding.” and that “part of that is also to optimise the structure in the bottom left, the R1/R2” and “you would infer that both those changes [R1/R2 and R] in different parts of the molecule were contributory, yes” [T2/1453-14611].
- The experts were also agreed that the ‘Further in vivo studies’ were in progress to optimise the PK properties of RD162, since there is no doubt that the most promising candidate identified in the Slides is RD162. In other words, the take home lead compound from the Slides (taking into account both activity and PK data) is RD162.
- Astellas also pointed out that RD162 shares the structure of RD37 with different substitutions being made to the right-hand phenyl ring (a methylamide group directly attached to the ring with an added fluorine on that phenyl ring). As with the other compound depicted in the Slides (namely, RD131), RD162 retains the cyclobutyl on the central ring, a feature of RD37.
- As before, what was in dispute was what the Skilled Team would do next, having read and considered the Slides with interest.
- For various reasons, the Claimants’ case of obviousness over the Slides is different (to that over the Poster). It is, of course, necessary to assess it as if the case over the Poster did not exist. So I start with a summary of the Claimants’ case on the Slides.
- At the relevant date there was a motivation to find treatments for HRPC. Applying Pozzoli:
i) The inventive concept of the Patent is that a compound with the structure of RD162’ has potential for treating prostate cancer or HRPC.
ii) The difference between that and the Slides is that compound of claim 1 (RD162’) has a dimethyl substituent at position X of the central ring whereas RD162 has a cyclobutyl substituent at that position;
iii) It would have been obvious to the Skilled Team that compounds with a similar structure to RD162 are likely to have similar activity, in particular it would have been obvious that a compound which is the same as RD162 but has a cyclopentyl or dimethyl substituent at position X would be likely to exhibit similar activity against hormone refractory cells.
iv) In the case of the Slides, the pharmacophore suggests that each of R1 and R2 could be methyl as these would be expected to behave similarly to the cyclobutyl group in RD162.
v) From that starting point, it would therefore have been obvious to the Skilled Medicinal Chemist that these specific variants of RD162 (including RD162’) would be likely to have the same or similar activity, and therefore also have therapeutic potential to treat prostate cancer.
- Once again, in closing, the Claimants’ case was more nuanced, and they emphasised the following points:
i) The benefits of the inclusion of the methyl amide and fluorine on the right-side phenyl ring will be apparent from Slide 16.
ii) The pharmacophore on Slide 8 specifically contemplates a dimethyl at position X. RU59063 with a dimethyl at position X is represented on the same page. The gem-dimethyl fits the pharmacophore: being a hydrocarbon which has the capacity to form hydrophobic interactions with AR:
‘The prediction would be a small change in a hydrophobic region would have, let us say, minimal impact on the overall activity profile. That remains to be proven by testing, yes.’ - XX Westwell T2/17715-18
iii) Had the authors intended to communicate that the dimethyl of RU59063 was to be avoided (and/or that cyclobutyl was materially better than the gem-dimethyl) they would not have drawn the pharmacophore the way they did.
- As before, I will first set out the context in which Prof Westwell formed his opinion that claim 1 was obvious based on the Slides.
- Having considered the disclosure of the Slides, the relevant context for his views on obviousness were set out in a new section entitled ‘Obvious steps to take in light of the Slides’. In the first paragraph of that section, he set out a similar three-fold reminder to that in [9.42]. In [10.28], he identified correctly the difference between the outcome from the Slides and claim 1 and then in [10.29] he used the same wording, mutatis mutandis, as in [9.47]. His reasons followed in [10.30]-[10.33]. Not surprisingly, these focussed on the pharmacophore shown in Slide 8.
- In summary, his reasons were that RD162 would be identified by the Skilled Medicinal Chemist as the most promising compound in the Slides since slide 16 showed it was the only compound identified as having antagonist activity in vitro combined with useful PK properties. He continued as follows:
‘10.30 ….. The skilled team would, according to the pharmacophore, expect certain variants to have the same or similar activity and therapeutic potential.
10.31 In terms of understanding the different parts of the molecule and their importance with respect to the performance, the skilled medicinal chemist would consult the pharmacophore from Slide 8. This explains that the left-hand side of the molecule is responsible for binding affinity to AR and therefore the skilled medicinal chemist would expect that modifications here would have the potential to have a significant impact on activity.
10.32 It also confirms that the rigid structure on the right-hand side of the molecule is required for antagonistic activity. Although certain changes to this part of the molecule are consistent with antagonistic activity in vitro (compare RD37, RD131 and RD162) it is only the inclusion of the methylamide substitution on the phenyl ring in combination with the fluorine substitution which gives rise to acceptable pharmacokinetic properties. The skilled medicinal chemist would not know whether such properties could be retained if that part of the molecule is altered.
10.33 According to the pharmacophore in Slide 8, the cyclobutyl in RD162 is designated R1/R2. The skilled medicinal chemist is informed that this part of the molecule is involved in hydrophobic interactions. The skilled medicinal chemist would want to keep R1 and R2 the same in order to avoid introducing an unwanted chiral centre, which I have explained in the CGK section. It would be obvious that each of R1 and R2 could be methyl as these would be expected to behave similarly to the cyclobutyl group in RD162 being only slightly smaller. They are also present in RU59063. The expectation would be that substituting two methyl groups for the cyclobutyl would not materially impact pharmacokinetic properties because they are both simply hydrocarbon groups. It follows that if RD162 is seen (as it would be) as having therapeutic potential it would be obvious to the skilled team on the basis of the data disclosed that a molecule in which the cyclobutyl has been replaced with dimethyl will be likely to have similar therapeutic potential. This would be obvious on the data disclosed in the Slides.’
- This reasoning is closely tied to material in the Slides. His 10.31 addresses the left-hand side and gives a reason why the Skilled Team would not want to make changes there.
- In 10.32 he addresses the right-hand side, making it clear that the phenyl ring must be retained but also reasons to retain (and not interfere with) the methylamide and fluorine substituents because it is those substituents which have provided the much-needed antagonist activity, particularly over the starting point of RU59063. By reference to the pharmacophore on Slide 8, Prof Westwell was effectively saying that the Skilled Team would not want to interfere with where the investigation of position X had reached.
- Then he turns to consider the only other area of investigation suggested in the pharmacophore - the R1 and R2 substituents at what I call position X. The pharmacophore shows very clearly that their role is ‘Hydrophobic interactions with AR’, as he says. He makes his point about wanting to avoid introducing a chiral centre by keeping R1 and R2 the same. Then he explains why, in his opinion, having R1 and R2 as methyl groups was obvious to the Skilled Team - I need not repeat his wording (set out above) because it is clear. It also seems entirely technically sensible. Indeed (and I will be corrected if I am wrong about this), the technical reasons he gave were not challenged.
- As before, Astellas’ challenge was that this was all driven by hindsight. Their principal point, as I understand it was that R1 and R2 were marked for investigation in the pharmacophore on Slide 8, by reference to RU59063 which featured the gem-dimethyl at position X. Although this is not stated in terms, they say the clear inference is that substituents at position X were investigated, and one can see the outcome - the adoption of the cyclobutyl in RD37 which is retained in RD131 and RD162.
- Astellas also attempted to characterise the Claimants’ case as nothing more than saying that because the pharmacophore shows that the group at R1 and R2 is involved in hydrophobic interactions, then it is obvious to try any hydrophobic substitution, including the dimethyl that has just been discarded (whilst at the same time, of course, using the specific right-hand group of RD162). This struck me as an exaggeration, but the issue remains as to whether it was obvious to investigate the dimethyl at position X.
- Astellas submitted that is not an obvious thing to do, relying on the following:
i) The only data in respect of the dimethyl is that it was used on the very agonist the Slides are trying to improve. In the CGK, it was also used on the agonist nilutamide.
ii) Any reader of the Slides can see that a deliberate decision was made to change the dimethyl. It is not then obvious to revert to it with no reason to do so.
iii) The reference to “hydrophobic interactions” necessarily implies a pocket (as explained by Prof Ward at [T4/4833-48416 & 48820-4895] and accepted by Prof Westwell in Westwell 1 §10.11). There is every reason to think from the CGK that the larger and slightly more hydrophobic cyclobutyl substituent provides a snugger fit with increased hydrophobic interactions than the smaller dimethyl seen in the agonist compounds (Ward T4/4849-16). This accords with the Conclusions on the Slides and Prof Westwell’s evidence that a skilled person would think that the change at R1 and R2 contributed to greater potency.
iv) A fair reading of the Slides is that the cyclobutyl better fits the hydrophobic pocket and assists in the improvement over the early dimethyl agonist compounds (Ward [T4/4876-11 & 48910-4905].
- In conclusion, Astellas submitted the Slides represent a disclosure which changes features of the existing art and says that the changes have brought benefits. The obviousness case is that it is obvious from this to change one of them back. Accordingly, Astellas submitted there is nothing obvious about it at all.
- The obviousness arguments in this case were finely balanced, the more so in relation to the Slides. I have changed my mind on obviousness over the Slides more than once because Prof Westwell’s technical reasons were cogent and because his approach (which I infer was formulated before he saw the Poster) was less clearly redolent of hindsight.
- The problem, once again, was that he did not explain the context in which his Skilled Team would make the considerations set out in his technical reasoning.
- In their cross-examination of Prof Ward, the Claimants strove to provide a context - the SAR starting from RD162 - in which they suggested the Skilled Team would have made and tested RD162’, as a byproduct of their characterisation of RD162. However, my findings in relation to the ‘obvious to do a SAR’ in the context of the Poster apply equally here.
- As I discussed above, there was a degree of exaggeration in the second, third and fourth points made by Astellas. After that exaggeration is stripped out, there remained some force in the underlying points. With those points in mind, the Skilled Team would be likely to regard a suggested change from cyclobutyl to dimethyl as something of a backward step. The advantage of the characterisation argument was that it provided a reason to ignore the fact that that step might be a backward one.
- In view of my reservations about certain parts of the evidence of Prof Ward, the characterisation argument came very close to succeeding but I have to bear in mind the following points:
i) First, that if the steps required to get from the prior art to the claim are obvious, it ought to be possible to explain that case clearly and in evidence in chief.
ii) Second, in the litigation process there is an intense focus and much analysis of the route(s) to obviousness and the obstacles in the way.
iii) Third, that it is hardly surprising that, with a skilful cross-examination driven by an intense focus on the target, an argument for obviousness may appear to have force.
- Naturally, I am not saying that an obviousness argument cannot be proved through cross-examination of the patentee’s expert witness. However, in the circumstances of this case, I am unable to conclude that I received sufficient primary evidence to establish the allegation of obviousness over the Slides.
- The Patent is entitled “Diarylhydantoin compound”. Astellas does not maintain any claims to priority and so the relevant date is the filing date of the Patent, 29 March 2006.
- The invention is said (at [0001]) to relate to “a diarylhydantoin compound and methods for synthesizing them and using them in the treatment of hormone refractory prostate cancer”. As both the medicinal chemistry experts point out, in fact the claimed compound is a diarylthiohydantoin (reflecting the presence of C=S rather than C=O), although nothing turns on this.
- At [0002] to [0007] the Patent provides some background to prostate cancer including its prevalence and existing treatment pathways. Bicalutamide is identified at [0004] as the most commonly used anti-androgen. Two weaknesses of current antiandrogens are identified: their weak antagonistic activity and their strong agonist activity when AR is overexpressed in HR prostate cancer. At [0008]-[0009] it is stated that there is a need for new thiohydantoin compounds and specifically selective modulators of the AR, such as modulators which are non-steroidal, non-toxic, and tissue selective.
- The invention is said to be the compound RD162’. The structure of RD162’ is given at [0011] and at [0010] it is said to have strong antagonistic activity with minimal agonistic activity against AR and to inhibit growth of HR prostate cancer. A method of synthesis of RD162’ is given at [0017].
- The detailed description includes an extensive section (at [0053]-[0217]) detailing the synthesis of diarylhydantoin and diarylthiohydantoin compounds. The synthesis of the claimed compound (RD162’) is described as Example 56 (at [0195]-[0201]). The closely related compounds RD162 and RD162’’ are described as Examples 52 and 57 respectively.
- The pharmacological examination of the compounds begins at [0218]. A series of assays are described and some (but not all) of the RD compounds are tested in various in vitro and in vivo models.
- At [0219] the compounds are said to have been subjected to tests using a human prostate cell line that had been engineered to over-express AR (LNCaP), which is described as showing features of HR prostate cancer.
- The antagonist activity of a number of RD compounds was measured using the method described at [0220]. The resulting IC50 values are reported in Table 1.
- The agonistic activity of those compounds was measured as described at [0222] and the results reported in Table 2. An alternative assay to measure agonist activity is described at [0226]. The results are reported in Table 3, and are said to be consistent with those reported in Table 2.
- The effect of some of the RD compounds on AR mitochondrial activity by MTS assay (said to be a surrogate for growth) was also described at [0228]-[0231]. The results are shown at Figs 3, 4 and 5. A second MTS study is described at [0234] in which hormone sensitive (rather than HR) LNCaP cells are used. Both RD7 and RD37 are said to inhibit the growth of these cells.
- The inhibitory effect on HR prostate cancer xenograft tumours is described at [0232]-[0233] by measuring the size of human-derived tumours implanted into immunocompromised mice. RD37 is said to have strongly inhibited the growth of LNCaP-AR derived and LAPC4 derived tumours in this model. In a similar experiment described at [0235], RD162 was reported to be effective to prevent tumour growth and even to cause shrinkage.
- A pharmacokinetic (PK) study on bicalutamide and three RD compounds (RD37, RD131 and RD162) is described at [0236]-[0240] and the results reported in Fig 13 and Table 4. Figures 15A and 15B also show these results. In addition to the IC50 and LogP values for RD131 and RD162, Table 4 reports the steady state plasma concentrations for bicalutamide, RD131 and RD162 in mice plasma.
- The description continues (from [0241]) by ranking the synthesised compounds into six tiers. The concept of ranking into tiers was introduced at [0054] in which it was said that the compounds in Tiers 1-3, 4 and 5-6 were expected to be “superior to”, “comparable to” and “worse than” bicalutamide respectively for the treatment of prostate cancer. At [0241] it is stated that the classification of compounds to particular tiers was based on consideration of the available in vitro, in vivo and PK data previously described as well as additional data not disclosed in the Patent. It is also explained that not all compounds were tested in each assay and a degree of judgment was applied in ranking the compounds relative to each other in terms of their expected utility in treating prostate cancer.
- The only data provided in the specification in respect of RD162’ is that at [0245] (in the discussion of the Tier 1 compounds) and Figure 21. The Claimants point out that:
i) RD162’ was not mentioned at all in any of the in vitro and in vivo studies described at [0218]-[0240].
ii) There is no PK data in respect of RD162’.
- Figure 21 is described (at [0043]) as “a graph representing PSA absorbance associated with LN-AR cells treated with various concentrations of RD162, RD162’, RD162’’, and RD170 and vehicle solution”. At [0245] it is stated that “Figure 21 shows that under treatment with RD162, RD162’, RD162", RD169, and RD170 at doses of 100, 200, 500, and 1000 nM, PSA levels of LN-AR cells decreased. Moreover, the higher the dose, the lower the PSA level.”
- Notwithstanding the terminology of ‘treatment’ and ‘dose’, it would be clear to the Skilled Team (not least from [0043]) that the data in fig.21 are from a cell-based assay rather than in vivo. The experts agree that fig. 21 shows the results of two different experiments.
- Fig. 21 (as supplemented by Prof Clarke) is shown below. I will briefly explain the coloured lines added by Prof Clarke in his reply report:
i) Lines in green identify where the lower bound of the RD162 result is higher than the upper bound of the RD162` result i.e. a concentration at which the performance of RD162` is better than RD162, and that that difference is meaningful, according to the method used by the patentee to generate the error bars.
ii) Lines in yellow identify an overlap between RD162 and RD162`, taking account of their error bars, giving the Skilled Cancer Biologist a lesser degree of certainty that any difference was meaningful.
iii) Lines in red denote results where the lower bound of the RD162` result is higher than the upper bound of the RD162 result i.e. a concentration at which the performance of RD162 is better than RD162` and that that difference is meaningful.

- There was a dispute between the experts as to what is disclosed by Fig. 21, which I discuss in the next section.
- From [0262]-[0266] the Patent sets out some commentary on the effect of various structural differences between compounds within the series and the ranking of those compounds within the various tiers.
- At [0267]-[0287] the Patent concludes with a lengthy high-level discussion of the various ways in which the claimed compound can be formulated and administered together with ranges for appropriate doses, none of which is relevant to the issues I have to decide.
- Not surprisingly, no issue of construction arises. Claim 1 claims a compound with the formula of RD162’ or a pharmaceutically acceptable salt thereof.
- In his first report, Prof Clarke’s view was that “the overall trend” shown in Fig. 21A “is a broadly consistent one, notably that RD162’ is more effective than RD162” and that in Fig. 21B “is one of apparent variability”. In his reply report, Prof Clarke was of the same view: on the basis of Figs. 21A and B, Prof Clarke’s conclusion was that the Skilled Cancer Biologist would consider it plausible that “RD162’ would be a more effective therapy than bicalutamide in treating HRPC” and that “at the higher or highest concentrations RD162' is likely to be more effective than RD162 as an antagonist of AR-overexpressed LNCaP cells and hence as a therapy for HRPC”.
- The Claimants pointed out that his latter conclusion was not one drawn by the authors of the Patent and characterised it as appearing at best, ‘strained’.
- Prof Hickson’s view (both in his first and second reports) was that based on the data in Figs. 21A and B “the Skilled Cancer Biologist could not say one way or another whether there was a difference in activity or likely performance as a potential prostate cancer therapeutic of RD162 and RD162' ”
- In their cross-examination of Prof Clarke, the Claimants attempted to paint the picture that he did not have the expertise to analyse the results of these assays because he had not personally conducted assays of this type or other tests used in this field. However, his answers in cross-examination firmly squashed this suggestion. He indicated he worked closely with his team and would regularly discuss the results of cell based testing with his Research Fellow - he could step out of his office and straight into that of his Research Fellow ‘so we had regular meetings and we had regular discussions… it was an integrated department, I was not actually putting the reagents in the dish but we were reviewing the results on a regular basis.’
- Professors Clarke and Hickson were addressing a rather narrow point on what the Skilled Team could deduce from the data provided in the Patent. The data in the Patent is by no means complete - not every compound is subjected to the same tests - but is consistent with certain tests being conducted at various stages to narrow the focus so as to identify the best molecule(s) to take forward to the next stage of the process.
- Thus, there was focus on Fig. 14, which compares bicalutamide, RD131 and RD162 against a control DMSO. There are some oddities but some are explained by the fact that the results have been normalised against the control DMSO (which is always 1). In his third report, Prof Hickson pointed out that this figure is using an absorbance scale where the data have been normalised and it was unusual to refer to ‘PSA Absorbance’ as ‘(fold)’.
- By far the greatest focus was on Fig.21. In the comparisons in Fig.21, Bicalutamide was not included. As can be seen, Fig.21A compares RD162, RD162’ and RD169 against a control (‘vehicle’), whereas Fig.21B compares RD162, RD162’, RD162” and RD170 against the control. The experts agreed that these were two different experiments likely conducted at different times.
- There was a good deal of discussion about the use of error bars in the various figures in the Patent (the significance of which is not explained) and whether the Skilled Team would be more interested in IC50 values or the maximum PSA absorbance (i.e. at the highest or higher concentrations of 500nM or 1000nM). However, as Prof Clarke indicated, we have to deal with the (incomplete) data provided.
- Ultimately, I got the impression that the error bars (and the disputes about them) were put on one side and the comparisons were made simply by the height of the histograms. Thus, in fig21A, there was no difference at the highest concentration between RD162 and RD162’, but at 500nM, RD162’ appears to be more effective than RD162. In fig.21B, by contrast, RD162’ is better than RD162 at both of the higher concentrations.
- In the end, despite his point that the Skilled Team would want to see IC50 data before drawing any conclusions but acknowledging those data are not provided (at least for RD162’), Prof Hickson appeared to agree with Prof Clarke that the Skilled Cancer Biologist would be interested at this stage in ‘maximal blockade’ and would want to identify a compound that is highly active. Prof Hickson agreed: ‘high potency at this stage would be indicative of an interesting compound’.
- Prof Clarke’s evidence on what a Skilled Cancer Biologist would take from Fig. 21 was:
Q. Nowhere do the inventors say that 162' may be or is superior to 162. It is not a conclusion that the inventors have drawn, is it?
A. But it is evident from graphs 21A and B to me that there is a signal that it is better. Clearly that would need further testing. What a skilled biologist, particularly somebody dealing with prostate cancer all the time, would be looking for is the best agent which produces the best anti-cancer effect in this setting.
- To the same effect, somewhat later he said:
I think one has to look at the
13 data as presented in this section of the patent and try to
14 work out which one looks to be the better agent, and which
15 might be the agent which one would take forward for further
16 testing.
17 Q. So you are saying that the result at 21B trumps the result at
18 21A; is that your approach?
19 A. No, it is not my approach. I think that there is evidence of
20 greater effect at the 500 nM dose in 21A, and there is
21 evidence of greater effect at the 1,000 nM dose in 21B. So,
22 in both of the separate sets of experiments, 162' has shown
23 superiority in areas. So, if one was to ask me, if you are
24 going to pick the winner amongst this lot, what would it be,
25 I would say, well, you are going to have to go with 162',
2 accepting fully that you would have to test for its
3 other properties” (emphasis added).
- In the light of the evidence, the criticisms levelled by the Claimants at Prof Clarke were either unfounded (re his experience and expertise) and/or they lost any real significance (re error bars).
- The Claimants alleged that the Patent does not plausibly disclose that there is any technical contribution in identifying RD162′ over the prior art. In particular it is said that the Slides and the Poster disclose a number of other compounds (referred to in the Patent as “Tier 1 compounds”), including RD162. It is alleged that there are no data in the Patent to show that RD162′ is efficacious in vivo or that its activity or suitability for treating prostate cancer is materially different (far less superior) to RD162.
- In their opening skeleton argument, the Claimants explained this plea as a squeeze between inventive step and technical contribution/sufficiency. Due to the way in which I have decided the obviousness allegations, no squeeze arises, but I propose to deal with the plea. The Claimants’ case was that there is no disclosure in the Patent that RD162’ has a materially different activity to RD162 or that the suitability of those two compounds for treating prostate cancer is materially different. Nor is such derivable from the application as filed ([A.1/2], which is, strictly speaking, the correct text from which to assess this issue: Sandoz v Bristol-Myers Squibb [2023] EWCA Civ 472 at [53]).
- Further, so the Claimants’ argument went, there is no in vivo data in the Patent relating to RD162’, and specifically there is no PK data for RD162’ showing that it has a PK profile that would make it a suitable drug candidate for treating prostate cancer.
- The Claimants contended that these points gave rise to an unavoidable squeeze with the obviousness case being run by Astellas. For it to be possible to make a reasonable prediction from the disclosure in the Patent/application that RD162’ will work to treat prostate cancer, the Claimants contended that the Skilled Team must be able to extrapolate from the limited PK data that the Patent/application provided for RD162. If that is the case, then the Claimants say it must follow that the Skilled Team would similarly be able to extrapolate from the data that the prior art provides for RD162 (and other RD compounds) to be able reasonably to predict the properties of a compound having the structure of RD162’. If such prediction is not possible then the Patent/application does not render plausible the treatment of hormone refractory prostate cancer or any other condition.
- By contrast, Astellas approached this issue purely as a plausibility issue and identified the following applicable principles.
- In Takeda v Roche [2019] EWHC 1911 (Pat); [2019] RPC 18, Birss J (as he then was) explained at [203] that the fact that the patent monopoly should be justified by the actual technical contribution to the art has often been referred to with approval in the UK, most recently by the Supreme Court in Warner-Lambert v Generics [2018] UKSC 56; [2019] 3 All ER 95 and in Actavis v ICOS [2019] UKSC 15; [2020] 1 All ER 213. Birss J went on to explain in Takeda at [204]:
One way in which this principle has been applied in the context of inventive step is to deny validity to a selection from the prior art “which is purely arbitrary and cannot be justified by some useful technical property”. Such a selection “is likely to be held to be obvious because it does not make a real technical advance”. These passages are taken from Floyd LJ in Generics UK Ltd t/a Mylan v Yeda [2013] EWCA Civ 925, citing Jacob LJ in Dr Reddy’s Laboratories (UK) Ltd v Eli Lilly and Co Ltd [2010] RPC 9.
- At [207] Birss J assessed a technical contribution over the prior art by asking the questions (a) is it disclosed in the Patent? (b) is it plausible? (c) is it true? (d) is it a technical advance? and (d) does it support claims of the breadth they are?
- In Generics v Lundbeck [2009] UKHL 12; [2009] 2 All ER 955 the claim was to a single enantiomer of citalopram. In the House of Lords the question was one of sufficiency and whether a technical contribution had been disclosed. At [83] Lord Neuberger explained:
‘It was also contended on behalf of the appellants that, if the Patent extended to escitalopram as a product, the respondents would be accorded a monopoly which exceeded their technical contribution to the art. Although it is an extra-statutory concept, I accept that, at least as a general rule, the monopoly to be granted to the patentee is to be assessed by reference to the “technical contribution” made by the teaching of the patent. That is an approach regularly adopted by the Technical Board of Appeal of the European Patent Office (“the Board”): see, for example, T409/91 EXXON/Fuel Oils [1994] OJEPO 653, para. 3.3. However, to put it at its lowest, it can be said that the Respondent’s technical contribution in this case was to make available, for the first time, a product which had previously been unavailable, namely the isolated (+)-enantiomer of citalopram. On that basis, it would appear to follow that the respondent was entitled to claim the enantiomer.’ (emphasis added)
- In Warner-Lambert v Generics [2018] UKSC 56; [2019] 3 All ER 95 Lord Sumption explained the approach in the UK to plausibility. The key propositions are as follows:
i) A patentee cannot claim a monopoly unless he not only makes but discloses a contribution to the art (at [35]).
ii) The disclosure in the patent must demonstrate in light of the CGK at the priority date that the claimed therapeutic effect is plausible (at [35]). The specification must disclose some reason for supposing that the implied assertion of efficacy in the claim is true. Plausibility is not a distinct condition of validity with a life of its own, but a standard against which that must be demonstrated (at [36]).
iii) The proposition that a product is efficacious for the treatment of a given condition is not made plausible by a bare assertion to that effect, and the disclosure of a mere possibility that it will work is not better than a bare assertion (at [37]).
iv) The claimed therapeutic effect may well be rendered plausible by a specification showing that something was worth trying for a reason, i.e. not just because there was an abstract possibility that it would work but because reasonable scientific grounds were disclosed for expecting that it might well work (at [37]).
v) Although the disclosure need not definitively prove the assertion that the product works for the designated purpose, there must be something that would cause the skilled person to think that there was a reasonable prospect that the assertion would prove to be true. This need not necessarily be demonstrated by experimental data. It can be demonstrated by a priori reasoning (at [37]).
vi) The question is not whether it works but whether the contribution to the art consisting of the discovery that it can be expected to work has been sufficiently disclosed in the patent (at [40]).
- In Sandoz & Teva v BMS [2022] EWHC 822 (Pat) (“Apixaban HC”) at [66], Meade J added that where the objection is of lack of plausibility in an AgrEvo-type situation, a patentee is not necessarily limited to the most demanding teaching of utility in the specification and is entitled to try to rely on a less ambitious degree of utility, or a utility of a different but related kind. The AgrEvo-type of objection is that there is no technical contribution at all and a patentee ought to be able to meet it by showing that there is some contribution even if it turns out that the contribution is less than the patentee thought.
- On appeal (“Apixaban CA”: [2023] EWCA Civ 472), Arnold LJ summarised the rule against “arbitrary” selections at [30]. At [31], he quoted (with approval) the judgment of Kitchin LJ (as he then was) in Idenix v Gilead [2016] EWCA Civ 1089 who described plausibility in the following terms: “There must be a real reason for supposing that the claimed invention will indeed have the promised technical effect”.
- Since Warner-Lambert, the Enlarged Board of the EPO in G 2/21 has considered plausibility and the so-called two lines of EPO case law (“ab initio plausibility” or “ab initio implausibility”). Arnold LJ explained at [47] that the Enlarged Board regarded the two lines of case law as being reconcilable. In each case, the core question being addressed was what the technical teaching of the application was to the skilled person with the CGK in mind at the filing date, and whether the technical effect relied upon by the patent applicant or proprietor was derivable from the application.
- The following further propositions can be derived from the Judgment in Apixaban CA:
i) Warner-Lambert remains binding, both for cases involving claims to second medical uses and to claims to a single chemical compound, and it was not suggested in that case that G 2/21 would justify the Court in departing from Warner-Lambert (at [91]-[92]).
ii) When considering inventive step, it is necessary to consider what technical problem the claimed invention solves. If it is not plausible that the invention solves any technical problem, then the patentee has made no technical contribution and the invention does not involve an inventive step. The same is true for sufficiency: if the disclosure does not make it plausible that the invention solves any technical problem, then the patentee has made no technical contribution and the specification does not disclose any invention (at [92]).
iii) The standard of plausibility to be applied is that of the majority in Warner-Lambert which corresponds to the “ab initio plausibility” test rather than the “ab initio implausibility” approach. The approach of the Enlarged Board appears much closer to the latter but it was not suggested that G 2/21 justifies departing from Warner-Lambert.
- As to what can amount to a solution to a technical problem, the EPO in AgrEvo made clear that a relevant technical contribution to the art can be the provision of further (alternative) compounds with certain activity or to treat a certain indication. The issue for the patentee in AgrEvo was that not all compounds covered by the (Markush formula) claim would plausibly have the claimed herbicidal activity. That meant that the technical contribution reduced merely to providing other compounds. That was not inventive (see AgrEvo at [2.6] quoted by Meade J in Apixaban HC at [28]-[29]).
- Finally, there is no requirement for an invention to have to be “better” than the prior art (and not a sensible way this could be evaluated). A new and non-obvious alternative solution to solve a problem remains patentable. This is made clear in a number of cases at the EPO, see for example the Case Law of the Boards of Appeal and decision T 588/93 where it makes clear that it is not necessary to show substantial or gradual improvement over the prior art, an invention can instead be an alternative solution to a known problem (see also T 179/108 at §12.5 and the Case Law book at §4.5).
- In closing, these issues of insufficiency/plausibility were dealt with relatively briefly and really only elucidated in oral closing submissions.
- For their part, the Claimants contended there was a genuine squeeze such that if they failed to establish obviousness, the Patent would lack plausibility in the absence of any PKDM testing or data on RD162’.
- The Claimants’ primary submission was designed to bolster their obviousness case. They contended that it was obvious from its similarity with the structure of RD162 and the data in Table 4 and Fig 15 that other compounds described in the Poster when combined with the methyl amide at position 4 and fluorine at position 3 would be ‘suitable for use’. This was the point stressed in the Claimants’ oral reply.
- The Claimants said they anticipated that Astellas would say that such predictions cannot be made on the basis of the Poster and contended that it therefore follows that they cannot be made on the basis of the Patent either. If Astellas are right, then the Claimants contended the necessary consequence was that the Patent is invalid because there is no data or reasoning which makes it plausible that RD162’ has PKDM properties which make it suitable for use.
- Astellas characterised the plausibility attack as being advanced in a very half-hearted way (if at all) in the Claimants’ opening. The pleaded attack on plausibility was explained in the opening skeleton as a squeeze on how far extrapolation to therapeutic use can be made in the absence of PK data in the Patent on RD162′. Very little was said about it in the oral opening, other than ‘……there is a squeeze, a potential squeeze, which is set out in our skeleton argument. We are not proposing to say any more about it at his stage. It is there. We can see where that ends up’.
- In closing, Astellas submitted there was no issue on plausibility, whether as a squeeze or otherwise, for the following reasons:
i) First, because both biologists agreed that RD162 is disclosed as being credibly better than bicalutamide [T2/798-12]. Both biologists thought that fig. 21 credibly shows that RD162′ has greater activity at the important highest concentrations than RD162 [T2/9420-9517 & T3/29719-2983 & T3/30316-21].
ii) Second, it was agreed CGK that a drug that decreases secreted PSA levels could potentially be useful as a treatment for prostate cancer (see [74] above). There was no dispute that the modified LNCaP cells used in fig. 21 of the Patent are suitable for assessing the ability of a compound to reduce PSA relevant to HR prostate cancer [T1/55-23].
- To address any alleged squeeze, Astellas submitted that the big difference between the Patent and the Poster or the Slides is that the Patent includes a head-to-head HR prostate cancer PSA absorbance assay between RD162 and RD162′ showing the potential improved activity of RD162′. The Poster and the Slides do not. The Poster shows the activity of RD7, but Prof Westwell accepted that it is not possible to translate those data over to RD162 to predict the activity of RD162′, because the substituents are different ([T2/18611-17]). It is not the structure of RD162′ which makes the claim to potential therapeutic use plausible: it is the head-to-head PSA absorbance data in fig. 21 - which both experts agreed rendered the claim credible or plausible. It is the data in fig. 21 that allows the Skilled Team to “pick the winner amongst this lot” [Clarke at T3/29719-2983]– that winner is RD162′.
- The parties had differing views of the problem addressed by the Patent and the technical contribution of it.
- Astellas’ position was that the Patent teaches a clear technical contribution over both the CGK and the cited prior art. It teaches a new molecule which plainly has good antagonistic activity in an HR prostate cancer PSA absorbance assay - a meaningful advance over bicalutamide. Astellas submit that is sufficient. But in this case, Astellas also submit the claimed compound is also plausibly more active than the preferred compound of the cited prior art.
- I outlined the Claimants’ position in opening and closing above. In their written closing, the Claimants’ proposition was that the Patent had to show that it was plausible that RD162’ had better PKDM properties than RD162, apparently seeking to exploit the fact that the Patent provides PK results for RD162 in fig.13 and Table 4, but no PK data for RD162’. I consider the Claimants, both in opening and closing, were focussing on the wrong question.
- Notwithstanding the volume of the submissions made on this allegation, in my judgment the answers are clear. First, the claim in the Patent to RD162’ is not based on some “bare assertion” or “abstract possibility” (as per Lord Sumption in Warner Lambert at [37]). Instead, in my judgment the reasons set out by Astellas in [439] above are clearly sufficient for the disclosure of the Patent plausibly to support the claims. After all, the Patent discloses and claims RD162’, a novel compound. It is plausible it will work and it does work. The provision of RD162’ is a technical advance. At the very least, RD162’ was a plausible alternative molecule to RD162, a point confirmed by its subsequent therapeutic use. So, in my judgment, the Patent plainly makes a technical contribution.
- Furthermore, as for the Claimants’ squeeze argument, it entails a comparison between:
i) Whether, on the data presented in the Poster or the Slides and the similarity in structure between RD162 and RD162’, it was plausible or possible to predict the activity of RD162’; as against
ii) Whether, on the data presented in the Patent, which included the cell-based assay comparison between RD162 and RD162’ in Fig 21, plus the patentee’s choice to place RD162’ in Tier 1, it was plausible that RD162’ would be suitable for therapeutic use.
- As explained in [440] above, there is no squeeze because the two elements of the supposed squeeze are not comparable. Furthermore, I have not rejected the obviousness attacks for any reason(s) connected to this sufficiency/plausibility issue, another reason why there is no squeeze.
- Accordingly, I dismiss the Insufficiency/Plausibility attack.
- Since I have found that claim 1 of EP196 was not obvious over either the Poster or the Slides and the insufficiency/plausibility attack also fails, I must dismiss each of the three actions brought by the Claimants.
