- This case concerns two patents which relate to a vaccine for respiratory syncytial virus (RSV). RSV is a common worldwide cause of lower respiratory tract infections in infants and young children, causing bronchiolitis and pneumonia. RSV is also an important cause of lower respiratory tract disease in the elderly and in people who are immunocompromised.
- This is also a case about so-called 'secondary evidence' of obviousness or lack of it. In particular it raises questions of whether the point needs to be pleaded and how it can and should be supported in evidence.
- The two patents in suit are EP (UK) 3 109 258 and EP (UK) 2 222 710 (the Patents, EP258 and EP710 respectively), both entitled "Recombinant RSV antigens". EP258 is a divisional application of EP710. The Patents describe a strategy for vaccinating against RSV. The strategy includes use of the F subunit vaccine stabilised in its prefusion conformation (sometimes referred to as "Pre Fusion" or "PreF"), and how to construct a stabilised F antigen in the prefusion conformation.
- The Patents are largely identical with the exception of the claims and the "Summary of the Invention". In EP 258, the key is it is a recombinant RSV polypeptide that is stabilised through use of a trimerisation domain, and in EP 710 the additional feature is the absence of furin cleavage sites. Both Patents claim priority from the same documents: US patent application 61/016,524 (filed on 24 December 2007 (the "Priority Date")) and US patent application 61/056, 206 (filed on 27 May 2008). Whether the Patents are entitled to the claimed priority is in issue in this action.
- The Defendants ('GSK') are the registered proprietor of the Patents. The Claimant ('Pfizer') say that the Patents are invalid and they sought to clear the way ahead of a commercial launch in the UK of their own RSV vaccine (known as "RSVPreF") for use in the prevention of RSV-associated disease.
- At the time of trial it was common ground there is no vaccine for RSV in the UK, but it has been a significant target for vaccine development for some years. Also at the time of trial, both parties had vaccines on the brink of obtaining approval in the UK. The GSK vaccine is for the elderly population only. The Pfizer RSVPreF, the subject of this claim, is for both elderly and the maternal population.
- GSK are not seeking an injunction in relation to the maternal indication, subject to suitable terms being agreed.
- The action as originally formulated was for revocation of three patents owned by GSK: EP710, EP258 and EP (UK) 3,178,490. They each concern recombinant (i.e., genetically engineered) RSV antigens. GSK has counterclaimed for infringement as a matter of 'normal' infringement and also under the doctrine of equivalents.
- GSK has submitted to judgment in respect of EP490 such that EP490 has been revoked and GSK has discontinued its counterclaim alleging infringement of the same. Accordingly, EP490 is no longer in issue in these proceedings.
- GSK now rely on claims 1, 5 and 8 (as proposed to be unconditionally amended) of EP258 and claims 1, 10, 22, 23 and 24 of EP710 as being independently valid.
- Critical issues at trial involved (a) the makeup of the Skilled Team (b) communication and collaboration between the members of the Team and (c) what was their collective CGK.
- Therefore the issues which I have to decide are:
i) The membership and skillsets of the Skilled Team.
ii) Their CGK.
iii) Claim interpretation. Two issues of interpretation arise on the claims: how to construe stabilizes and the meaning of the term polypeptide.
iv) Infringement. GSK alleges that Pfizer's product, RSVPreF, infringes each of EP 258 and EP 710. RSVPreF contains RSV F antigens which, the PPD accepts, are in the prefusion form. Infringement is put on the basis of normal infringement and infringement by equivalence.
v) Priority. There is a formal challenge to priority of both Patents which gives rise to issues on Belgian law. If that is successful then WO456 becomes full prior art for inventive step, otherwise it is a novelty only citation. The Belgian law issues are self-contained and it is convenient to deal with those in a separate section. I have, however, applied the result of my analysis when considering WO456.
vi) Novelty at the priority date. WO456 is intervening novelty-only art at the priority date and is said to anticipate claims 1 and 2 of EP 258.
vii) Obviousness at the priority date. The art cited against both Patents are Yin, the Jardetzky disclosures, and the ASV Abstract. In response, GSK developed, very largely in cross-examination, a case on secondary evidence of non-obviousness.
viii) Obviousness at the filing date if priority is lost. Pfizer relies on WO 456 as full art (in addition to Yin, the Jardetzky disclosures, and the ASV Abstract) against both Patents.
ix) AgrEvo obviousness. Pfizer only relies on this as a squeeze on GSK's construction of the term stabilizes.
x) Insufficiency. Pfizer's insufficiency case is limited to a single point that engages matters of undue burden and uncertainty, together with an enablement squeeze over the prior art, and as a plausibility attack in respect of the use of a product claimed without an adjuvant (this is aimed at certain claims only).
xi) Arrow relief. Pfizer also seeks Arrow relief declaring that at the priority and/or filing date it was obvious to make an antigen with certain features of RSV PreF and to use it in the treatment or prevention of RSV-associated diseases.
- Although all of these issues are important, the most significant battleground at trial concerned the first two. The disputes over what each piece of prior art disclosed were relatively minor, in comparison to the central disputes over the makeup of the Skilled Team and their resulting CGK.
- Anticipating the findings I make later in this Judgment, there were three major decision points in this case:
i) The first is as to the makeup of the Skilled Team and their skillsets. In short, did the Skilled Team include a Skilled Structural Biologist or was the molecular biology knowledge of the Skilled Vaccinologist sufficient?
ii) The decision on that first point has a major effect on the scope of the CGK, but did not resolve all the disputes as to what was CGK, which is the second major decision point. Particular pieces of knowledge, said to be CGK, formed the essential foundation to Pfizer's obviousness allegations. These points were also engaged by GSK's case on secondary evidence, so it is not possible to resolve the CGK disputes until a relatively late stage in this judgment, once I have considered the secondary evidence.
iii) The third major decision point concerns the allegations of obviousness. This is a case where the Skilled Team's reaction to each piece of prior art is extremely dependent on the CGK. Pfizer's case was that the CGK provided very fertile ground against which the disclosure of each piece of prior art must be viewed. GSK fundamentally disagreed. Again, it is not possible to reach final conclusions without considering GSK's case on secondary evidence.
- I received evidence of fact from Professor Theodore Jardetzky and Mr Michael Gilbert.
- Professor Jardetzky is a Professor in the Department of Structural Biology at Stanford University School of Medicine in California, USA, a position he took up in August/September 2007. He has more than 30 years of experience in the field of structural biology and studies the structures and mechanisms of macromolecular complexes important in viral pathogenesis. His laboratory has solved the structures of key entry glycoproteins involved in the cell targeting and membrane fusion steps of paramyxovirus, including the fusion (F) glycoprotein, and in close collaborations with virology groups, conducted many functional studies of these proteins.
- He gave evidence about a presentation he gave at the 6th International Respiratory Syncytial Virus Symposium ("RSV 2007 Symposium") which took place in October 2007. Professor Jardetzky and his group are responsible for much of the documentary prior art relied on in this case (Yin, the Jardetzky Abstract and the Jardetzky Slides). However the purpose of his evidence concerned what he said to the assembled audience when giving his presentation which included the Jardetzky Slides.
- I discuss the challenges made by GSK to Professor Jardetzky's evidence and his recollection below. Generally, I was satisfied he was genuinely assisting the Court and gave his evidence entirely honestly and fairly.
- Mr Gilbert is a partner at Marks & Clerk Law LLP, solicitors for Pfizer. He gave evidence in relation to Pfizer's claim for Arrow relief and, in particular, as to the importance of RSV vaccines in the UK and the various divisional applications made by GSK which remained outstanding. He also gave a further witness statement addressing an issue of disclosure that had arisen which related to the presentation given by Prof Jardetzky at the RSV Symposium. As expected from a solicitor of a representative firm, Mr Gilbert gave his evidence carefully and robustly.
- Each side called two technical experts plus an expert in Belgian Law. The issues of Belgian Law are separate and it is convenient to address both the witnesses and the issues in a separate section below. Pfizer called Dr Teresa Johnson as their principal expert to deal with vaccinology and Professor Winfried Weissenhorn to give evidence on structural biology. GSK called Dr Geraldine Taylor as their vaccinologist and Professor Anthony Wilkinson on molecular/structural biology matters. Here I give brief details of their relevant experience and provide my general views of their evidence.
- Dr Johnson has over 28 years of experience in the study of immunology and vaccinology, including in the context of RSV. From 2000 to 2011 she worked at the National Institute of Health's Vaccine Research Center under Dr Barney Graham. Dr Johnson is currently the Discipline Chair for Microbiology and Immunology at Edward Via College of Osteopathic Medicine in Virginia, USA.
- In their closing submissions, GSK made no criticism of the way Dr Johnson gave evidence and they accepted she had done her best to assist the Court. Their point was her evidence was tainted with hindsight and levelled particular criticism at the way in which her first report was prepared and her evidence on the CGK. I discuss both these matters below.
- Generally, however, Dr Johnson was a valuable witness who was plainly doing her best to assist the Court from a position of independence.
- Prof Weissenhorn has over three decades of experience in structural and molecular biology, particularly in relation to structure determination of proteins and complexes within the field of structural biology and host-pathogen interaction, including viral glycoproteins. He is currently Professor of Structural Biology and Biochemistry at the University Grenoble Alpes.
- GSK again had no criticisms of Professor Weissenhorn as a witness although they contended he was 'rather literalist' on the meaning of 'polypeptide'. GSK were also critical of the way in which the CGK sections of this report were prepared, but I have taken those points into consideration in my findings as to CGK below.
- Professor Weissenhorn was a fair witness. He answered the questions put to him and did his best to assist the court. He has first-hand experience of working on glycoprotein class I fusion protein vaccines, in HIV. He had read both the Yin papers at the priority date. As I explain in more detail below, the skill set, CGK and input to the skilled team in Prof Weissenhorn's evidence, which was correctly taken into account by Dr Johnson, is reflective of and consistent with that of real world teams at the priority date, and indeed with the personal experience of Dr Taylor.
- Dr Taylor's research on RSV immunology and RSV vaccines began in the early 1980, so she has a long career in the field. She is an honorary fellow at The Pirbright Institute and in 2007 was appointed a Jenner Investigator at the Jenner Institute, leading a vaccine programme employing post-docs, research assistants and doctoral students. She has taught on the Human and Veterinary Vaccinology course at Oxford University.
- Unfortunately, I need to resolve a number of issues regarding her evidence, which are interrelated. First, the way she was instructed, which had a profound influence on her written evidence. Second, some features of her cross-examination, in respect of which GSK made a number of criticisms. Third, the rather extensive 'corrections' to the transcript of the first day of her cross-examination, made at the start of the second day. I address these points in a later section.
- He is a Professor in the Department of Chemistry at York University and the current Head of the York Structural Biology Laboratory.
- I did not understand Pfizer to make any criticism of him personally or the way he gave his evidence in the witness box. However, Pfizer were highly critical of his selection as a witness and the way in which he was instructed. Pfizer established in cross-examination that he had no personal knowledge of how vaccine research groups that might or might not have been interested in RSV or other paramyxoviridae were operating around the priority date, that he had not been involved in vaccine research and had not worked on any subunit vaccine.
- As to the way in which he was instructed, Pfizer had three main criticisms: first, of the 'siloed' approach taken to the preparation of GSK's evidence; second, the fact that basic concepts such as CGK were never explained to him and third, his evidence relating to the meaning of the term 'polypeptide'. On the latter point, Pfizer contended that Professor Wilkinson's approach was based on the particular approach of his own structural biology lab, the York Structural Biology Laboratory, without taking into account the views or approach of any other member of the Skilled Team. To the extent necessary and justified, I have taken all these criticisms into account, particularly in relation to the interpretation of 'polypeptide'.
- It is necessary to consider how certain issues developed in this case and certain criticisms of what occurred during the evidence. This is conveniently done once I have determined the issue over the makeup of the Skilled Team and set out what the parties managed to agree was CGK.
- Before I proceed further, I consider it helpful to consider the principal contentions made by GSK in their closing submissions because they engage several of the key issues I have to decide concerning the Skilled Team, their CGK and the allegations of obviousness.
- At trial, GSK's response to Pfizer's case on obviousness was developed in two parts and I should introduce the two parts here because they affect the major decision points I have already outlined.
- On the primary evidence of obviousness, GSK made an overarching submission. It was GSK's case that to arrive at the invention, the Skilled Team had to take the following six steps:
(1) First, the skilled vaccinologist had to decide to revisit F subunit vaccines in preference to what GSK alleged were the more favoured approaches.
(2) Second, the skilled vaccinologist had to decide that the preferred alternative approach to the subunit F vaccine is revisiting its structure rather than the manner of its presentation (adjuvants, modes of delivery etc.).
(3) Third the skilled vaccinologist had to recognise that problem with existing F subunit vaccines, such as PFP, was that they were in the postfusion conformation, a problem not previously recognised in the literature.
(4) Fourth the skilled vaccinologist had to consider that it might be possible to address this issue by making an alternative form of F subunit antigen, as opposed to just abandoning subunits in favour of live attenuated vaccines, vectored vaccines, or DNA vaccines.
(5) Fifth, the skilled team had to pursue an F subunit antigen in the prefusion form as opposed to the intermediate form or some other form.
(6) The Sixth step that needed to be taken was that the skilled team needed to find a source of the prefusion F protein, something which GSK contended was not readily available because a stabilised form of prefusion F protein had never been made before. So the skilled team (without the benefit of the teaching in the Patents) needed to pursue a collaboration with a skilled structural biologist to determine for the first time whether a soluble F protein of RSV could be stabilised in the prefusion conformation. GSK pointed out that Professor Jardetzky said he was unable to achieve this when he tried. GSK contended this step was not trivial and would have required the skilled team to be sufficiently motivated.
- Two immediate points may be noted:
i) First, none of these six steps engages any of the cited prior art. Thus, GSK appeared to be responding to a case of obviousness over the CGK alone, which was not a case which Pfizer was running.
ii) Second, on GSK's case, the skilled structural biologist is only brought in at the Sixth step.
- Furthermore, GSK levelled the following principal criticisms at Pfizer's case:
i) First, that Pfizer had re-defined the Skilled Team in the course of their cross-examination of Dr Taylor to include the 'Melero-type structural virologist'.
ii) Second, GSK alleged that Pfizer had failed to define the CGK properly. The nub of this criticism appeared to rest on two propositions: first, that to rely on memory alone was inadequate, the corollary being that it was necessary to find every proposition said to be CGK set out clearly in a pre-priority textbook or scientific paper; and second, that the individual recollection of Dr Johnson was not representative of what was generally known.
iii) Third, GSK alleged that Pfizer's case was essentially one of obviousness over CGK. GSK's points under this head were that (1) the key reasoning in Pfizer's obviousness cases was not found in the prior art; (2) that it was important to distinguish between the CGK and what is said to be obvious in the light of the CGK; (3) the cross-examination jumped from one passage to another, ignoring inconvenient detail and context; and (4) the reliance in particular on the alleged CGK proposition that antigens should be in their 'native' conformation.
iv) GSK's fourth point was specific to Pfizer's case of insufficiency and is best considered in that context.
v) Fifth, but separately, GSK criticised Pfizer's approach to the Skilled Team, contending that Pfizer approached the Skilled Team from the starting point of the solution in the Patents rather than the problem facing the Team in the art. Of course, Illumina question 1 proceeds on the basis of the problem.
- At trial, in opposition to Pfizer's allegations of obviousness, GSK sought to establish their case on secondary evidence in the cross-examination of Dr Johnson, the expert in vaccinology called by Pfizer. This case featured heavily in its closing submissions, in conjunction with the limited evidence from their vaccinology expert, Dr Taylor, on the point.
- I should add that GSK's case on secondary evidence was developed in a highly unsatisfactory manner. There was no pleading to foreshadow it, and the case as finally developed was hardly developed at all in Dr Taylor's evidence. Instead, two large bundles of technical papers and textbook extracts were agreed and prepared for trial, along with further technical papers in the cross-examination bundles served for Drs Taylor and Johnson. GSK's case on secondary evidence was really only fully explained in their written closing submissions, based primarily on extensive cross-examination of Dr Johnson on a variety of post-priority publications in conjunction with the limited evidence from their vaccinology expert, Dr Taylor, on the point. Most of these publications had not been discussed in the expert evidence, so on some key papers I was left to decide technical disputes without the assistance of any expert evidence.
- Although the experts exhibited certain chapters from the two key textbooks and a number of scientific papers, the cross-examination was conducted by reference to what amounted to three or four large bundles of dense scientific material. These materials reflected the fact that the Skilled Team in this case required knowledge in a number of disciplines but also that real-life teams conducting RSV research were highly knowledgeable and skilled. They were involved in cutting-edge science.
- There was a degree of common ground as to the makeup of the Skilled Team. Both sides agreed that the Team would be led by a vaccinologist and the Team would have expertise in immunology, virology and structural/molecular biology. However, GSK contended that the vaccinologist would have sufficient knowledge of molecular biology such that s/he would not consult a specialist in structural/molecular biology. What was really in dispute was the level of common general knowledge in the Skilled Team relating to matters of protein structures - i.e. the expertise or the extent of the skillset of what I will refer to as the Skilled Structural Biologist.
- I can clear away one issue immediately which involved a minor dispute as to the description of this 'structural biologist'. Other terms used in the evidence and at trial were 'molecular biologist' and 'structural virologist', but in my view all these terms were being used to describe the same member of the Skilled Team and at the same and correct level of generality.
- GSK's case was that the advanced knowledge of a specialist structural biologist was not required and that the lower level of knowledge possessed by a vaccinologist such as Dr Taylor was sufficient. Pfizer contended that a specialist would be consulted and be a member of the Skilled Team.
- As noted above, the Patents concern F subunit vaccines stabilised in the PreFusion or PreF conformation. As explained in greater detail below, it is important to stabilise the protein in the PreFusion conformation because without stabilisation this conformation is metastable and readily transforms into its PostFusion conformation which, as a vaccine, is ineffective. This brief introduction explains why the structure of the protein is critical.
- Accordingly, the issues I have to decide concerning the Skilled Team are as follows:
i) The level of expertise/extent of skillset of both the vaccinologist and the structural/molecular biologist.
ii) How the skilled vaccinologist and structural biologist work together and interact with each other. In this regard, Pfizer said that GSK's experts had been unduly "siloed" from one another and prevented from sharing their views as a real skilled team would.
- There was no real dispute as the applicable principles. The skilled addressee of each Patent is a person with a practical interest in the subject matter of the patent under consideration, possessed of the common general knowledge, and diligent but uninventive/unimaginative.
- The parties cited familiar authorities on the correct approach to this issue. I have addressed it in recent cases, by reference to my decision in Alcon Eye Care UK Ltd v AMO Development, LLC [2022] EWHC 955 (Pat) drawing on the principles concerning the identification of the skilled person or team, as set out by Henry Carr J. in Garmin (Europe) Limited v Koninklijke Philips N.V. [2019] EWHC 107 (Pat) and the decision of Birss J, as he then was, in Illumina v. Latvia [2021] EWHC 57 (Pat).
- GSK made some criticism of Pfizer's approach as failing to follow the approach described in Illumina in that (so it is alleged - see the Fifth point above) it starts from the solution in the Patents and not the problem in the established field to assemble its skilled team. Little turns on this in any event as I will proceed by reference to the three questions posed by Birss J in Illumina at [68].
- Prior to the closing submissions, there was the occasional hint that that GSK might be contending that this was a Schlumberger-type case, but the point was not pursued. In any event, I am satisfied that the skilled team to whom the Patents are addressed, and for considering sufficiency, is the same as that for the purposes of considering obviousness. The evidence established that there was an established field, in which real teams operated.
- However, that final point of criticism embodies an allegation of hindsight in putting together Pfizer's Skilled Team. The advantage of the Illumina questions is that they guard against a hindsight approach.
- It seems to me that another way of guarding against a hindsight approach in this case is to address at the outset what the undisputed member of the Skilled Team (i.e. the vaccinologist) would do having read a cited piece of prior art. I can take Yin by way of example. Although I am anticipating the more detailed analysis of Yin which I set out later in this judgment, it is clear, in my judgment that the skilled vaccinologist, having read Yin with interest would immediately recognise (a) the relevance of the structural analysis of the pre and post fusion conformations of PIV to RSV; and (b) the relevance of being able to stabilise the prefusion conformation of F with GCNt, but would also immediately call upon the skilled structural biologist to be a member of the team to take Yin forward.
- This analysis is entirely in line with the evidence of certain real life pre-priority collaborations where vaccinologists called upon the more detailed structural knowledge of structural biologists. This did not occur in every project aimed at trying to develop an RSV vaccine, only those where a structural issue arose.
- It is relevant also to keep in mind some evidence which Dr Johnson gave about the RSV field. Some interesting figures were put to her as to the amount of funding devoted to research into various viruses between 2000-2009. There was a very striking contrast between the funding for HIV and RSV. RSV was a much smaller field, relatively speaking. It was also a small field in absolute terms, despite the importance of a vaccine for RSV. I got the impression from Dr Johnson's evidence that everyone in the RSV field knew everyone else. The field comprised of groups working on developing RSV vaccines but was not confined to such groups. It seems there were also groups undertaking pure scientific research and/or research into vaccines for related viruses which was of relevance to those in the RSV field. I have in mind research relating to other paramyxoviruses but also other class 1 fusion viruses, even though I entirely accept that the Skilled Team could not keep up to date with all of the massive numbers of papers published in the HIV field.
- Furthermore, Dr Johnson gave some important evidence about how teams in the RSV field were funded and how they operated. Although she worked at the Vaccine Research Center at the National Institute of Health between 2000-2011 and did not have to secure grant funding in the same way as groups outside the NIH, I am sure projects at the VRC still competed for resources. So, a team which wished to conduct research into an RSV vaccine would have to put together a project proposal, have it approved, assemble the relevant team and conduct the project. It was apparent from the evidence that these projects continued for some years. Importantly, it was clear from the evidence that a group would not simply abandon a project mid-stream unless, of course, results were obtained which put the whole rationale for the project in doubt.
- As such, real-life teams were engaged in specific projects. They were not sitting around ready to pick up suggestions made in prior art.
- With that background, I can address the specific Illumina questions.
- First, what problem does the Patent seek to solve? In my view the problem it seeks to solve is developing a recombinant RSV F antigen, for use as a vaccine.
- Second, what was the established field in which the problem was located? This includes consideration of real teams. In a broad sense the answer is vaccine development, in particular a team that was interested in developing an RSV vaccine and setting the strategic course.
- Pfizer's case was that to work on the development of vaccine antigens required consideration of how modifications to the protein could affect its structure. The skilled team would therefore require someone with knowledge of the structure and function of RSV F necessary for the design of any subunit and for investigating conformational epitopes.
- GSK's case was that advanced structural biology was not necessary to put the invention into effect, the level of knowledge regarding standard molecular biology techniques possessed by someone like Dr Taylor was enough.
- Dr Taylor cited her own team at Pirbright as being typical in terms of skillset and organisation of a team interested in the development of HRSV vaccines at the priority date. Her team included a molecular biologist who she described as 'a skilled and experienced technician, trained in both molecular biology and virology'. Dr Taylor relied on this person to make their experimental constructs.
- In response, Professor Weissenhorn said that even if the person carrying out the molecular manipulation was an 'experienced technician' whose job was to focus on producing the proteins, there would need to be someone within the team who was considering the implications of those changes on the structure and function of the antigen. He said that could come from a structural biologist or a virologist or some other member and added that the title is not necessarily important but the skill set is.
- Professor Weissenhorn also said this skillset was reflected by teams working on viral fusion proteins at the time. He gave several examples of leading virologists such as John Skehel (influenza), Dennis Burton (HIV), Joseph Sodroski (HIV), Bob Lamb (paramyxoviruses) and John Moore (HIV) who were classified as virologists but had broad expertise and interests encompassing at least immunology, molecular biology and structural biology. He also mentioned José Melero as a well-known virologist in the RSV field, who had published papers with Skehel and Wiley in the early 2000s. In this reports, he said that many of those virologists also worked closely with structural biologists such as Don Wiley, Peter Kwong, Ian Wilson, Bob Lamb (who worked closely with Ted Jardetzky) and Peter Kim (who he characterised as a biochemist/biophysicist with ample expertise and knowledge of structural biology of viral envelope proteins and whose lab developed the isoleucine zipper and published the first crystal structure of the fusion core of RSV F in 2000).
- I note in particular the collaborations which were mentioned in evidence between Dr Taylor and Dr Wertz, in which it was Dr Wertz who made the recombinant construct for human RSV. Similarly, Dr Taylor provided evidence of collaborations with Dr Melero who was responsible for the structural matters on such projects. Dr Johnson also gave evidence of certain collaborations with structural specialists.
- Accordingly, the evidence established that real world vaccinology teams would typically have a team member with expertise in the structural biology of the virus under consideration, depending on the extent to which structural matters affected the antigen the Team is assessing or trying to create. Investigations in immunology would consider the structure of the epitope being investigated.
- It is clear that not every RSV vaccine project required a structural specialist. However, there were, on reflection, numerous indicators in this case that a Skilled Structural Biologist was a necessary member of the Skilled Team:
i) First, I reached the clear view that on reading and considering each piece of prior art in this case, the Skilled Vaccinologist would call upon and incorporate into the Skilled Team a Skilled Structural Biologist.
ii) Furthermore, in order to implement either of the Patents, the Skilled Vaccinologist would do the same. In both cases, the critical teaching lies in the protein structures. 'Ordinary' molecular biology knowledge at the level suggested by Dr Taylor would not be sufficient. In my judgment, the Skilled Team required a team member with more advanced structural biology expertise.
iii) Both these points were confirmed once I had understood that Dr Taylor's view of the CGK and the Statement of Agreed CGK omitted some critical points which were essential to understand the relevant technology in this case.
- Consideration of the real-world teams working on subunit RSV vaccines also demonstrated that they were true collaborations. In his written evidence, Professor Wilkinson envisaged a one-way flow of information with the vaccinologist posing specific questions to the structural biologist, although in cross-examination he ultimately agreed that it would be a two-way discussion.
- The other point taken by Professor Wilkinson was that he was not aware that structural biologists were designing vaccines at the priority date. His point rather confirms the siloed approach on GSK's side. It also misses the point, which is that the members of the team would collaborate and discuss the way(s) forward, having considered the relevant piece of prior art.
- I therefore answer the third question as follows: the skilled team would include a skilled vaccinologist and a structural biologist with advanced expertise in structural biology.
- As to whether GSK siloed its experts in an inappropriate manner, it is worth reiterating that members of real teams will communicate and the process of evidence preparation in patent cases ought to allow this to be reflected.
- In the present case, I agree that GSK adopted a siloed approach to the expertise of its different members of the skilled team which meant that Dr Taylor approached the CGK and prior art in a manner which was unduly narrow. This led to Dr Taylor disregarding most of the prior art as she said there was nothing of interest in it because it was not specifically focused on RSV. Further, this approach allowed Professor Wilkinson not to address it at all. This is not a criticism of the experts themselves, rather a criticism of the manner in which they were instructed.
- Whilst GSK also suggested that Pfizer's experts did not consult each other, there was no cross-examination to further substantiate this point.
- Whilst the cross-examination of Dr Taylor and the evidence that emerged provided the Court with insight as to the knowledge and characteristics of the Skilled Team, the written reports of Dr Taylor were insufficient on the point. I therefore agree with Pfizer that the partitioning of the CGK has been unhelpful and, crucially, unrealistic.
- In light of the above discussion, the approach of the team is a collaborative process between the skilled vaccinologist and the skilled structural biologist.
- That leaves the final point as to the skillset of the vaccinologist which I have already foreshadowed. As indicated, this issue arose due to the narrow approach taken by Dr Taylor to the prior art. I have to discuss her evidence in greater detail below, but in her written evidence she dismissed the Jardetzky and Yin prior art on the basis that neither concerned RSV. All that prior art was concerned with other members of the Paramyxovirus family, namely parainfluenza viruses (PIV). The evidence established to my complete satisfaction that the skilled vaccinologist would know about and would consider developments in structurally related viruses, including PIV. I have to discuss the extent of this cross-fertilisation to resolve certain of the disputes over CGK. However, it will be apparent that Dr Taylor's narrow approach in her written evidence had an impact on what could be agreed as being CGK.
- This section is entitled 'Technical Background' because its contents are required to understand the disputes over what was Common General Knowledge, which I must address later. However, anticipating the decisions I make later in this Judgment on those disputes, it will be seen that in fact I find that the whole of this section represents the CGK of the Skilled Team.
- At my request the parties provided a Statement of Agreed CGK. This was a helpful introduction to the technology, and I am very grateful for the work done in preparing that document. Once so introduced, the usefulness of the document changed because, on further analysis, the Statement of Agreed CGK appeared somewhat disjointed and incomplete. This section is expressed in the present tense, but represents the situation as at the Priority Date.
- The vaccinology parts were principally based on paragraphs from Dr Johnson's first report, supplemented by certain paragraphs from Dr Taylor. For the Molecular/Structural Biology section, this was based purely on extracts from Professor Weissenhorn's first report, with a few additions from his third report where he addressed certain points made by Professor Wilkinson. As was made clear in cross-examination, Professor Wilkinson was not asked to address CGK, indeed the concept was not even explained to him, although in his reply report, he disputed certain matters said to be CGK by Professor Weissenhorn, and I have considered his points when compiling the parts on structural aspects. However, the paragraphs from the CGK sections of Dr Johnson and Professor Weissenhorn's reports which were not included in the Statement of Agreed CGK served to highlight the CGK points in dispute and the issue over the Skilled Team which I have already addressed. They also explained why the Agreed Statement appeared disjointed and incomplete.
- What follows is based on the Statement of Agreed CGK, with various additional paragraphs and some re-ordering. Now that I have resolved the issues over the composition and skillsets of the Skilled Team, it is necessary to add in a number of paragraphs which were excluded due to those issues.
- It is convenient to remind myself here of the applicable legal principles, on which there was no dispute, with the relevant law set out in KCI Licencing Inc v Smith & Nephew plc [2010] EWHC 1487 (Pat), [2010] FSR 31 at [105]-[112]. By way of summary, in order to form part of the CGK, information must be generally known in the art, and regarded as a good basis for future action. Material that would be found by routine research in the course of developing the cited prior art may be taken into account in assessing obviousness, but it is not CGK as such.
A summary of the CGK disputes
- By the time of Closing Submissions, there remained a considerable dispute over the CGK, but I can record here two points which were common ground:
i) First, that subunit vaccines were a known strategy for RSV vaccination at the priority date, although there was a dispute as to their status.
ii) Second, that two key targets for RSV vaccination were F and G proteins.
- The principal disputes concerned the following topics which I introduce briefly here:
i) The extent to which adjuvants were necessary or generally used or expected to be necessary in subunit vaccines.
ii) Whether RSV vaccinologists were considering the different conformations of the RSV F protein in their approach to vaccine design.
iii) The relevance of researching recombinant subunit vaccines for related viruses to RSV which share the Class I fusion protein mechanism known to be a main target for RSV vaccines, including other paramyxoviruses, and HIV-1 and influenza HA.
iv) Whether references in the papers to the "natural", "mature" or "native" F protein would be understood by the skilled vaccinologist as the "prefusion" form.
v) Whether it was known that the most effective neutralizing antibodies would be likely to bind to the prefusion conformation of the F protein.
vi) Whether the skilled team had an awareness of and expertise in stabilising fusion glycoproteins, and that this was important for structural studies and for immunization experiments in a vaccine context.
- It is common ground that the two textbooks, Fields Virology ("Fields") (5th Edition, 2007) and The Respiratory Syncytial Virus (edited by Patricia Cane (2006) ("Cane"), were sources of CGK and that their contents were reflective of the CGK of the skilled vaccinologist at the Priority Date. It is clear that the account in Fields was more up to date and included some important recent developments (including, in particular, Yin).
- Before addressing some basic concepts of immunology and vaccinology, it is necessary to start with some basics of molecular biology.
Proteins
- Proteins are the molecules that provide many of the structures and machinery required to make cells (and viruses) work. Proteins are made of amino acids assembling into small or large or complex macromolecules alone or with co-factors.
- Amino acids have a carboxyl group and an amino group bonded to the same carbon atom, known as the α carbon. Amino acids are the building blocks of proteins and acids join together to form dipeptides, tripeptides, polypeptides, which are the substance of proteins, by use of a peptide bond. A peptide bond is formed between the α-nitrogen atom of one amino acid and the α -carboxyl group of a second amino acid in a condensation reaction, with the loss of one molecule of water. See Figure 10.
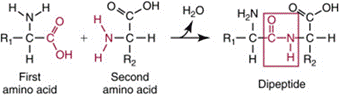
Figure 10: Diagram of a peptide bond between two amino acids
- A polypeptide is made up of a chain of amino acids joined by a type of covalent bond called peptide (or amide) bonds. Once a polypeptide is folded and becomes functional, the polypeptide is called a protein. Although many proteins consist of a single polypeptide, some are made up of multiple polypeptides which can be held together by hydrophobic interactions, hydrogen bonds, salt bridges and another type of covalent bond called disulphide bridges.
- A domain is a functional unit within a protein, and it can be on the same or different polypeptides in any one protein. A domain is the part of a protein that has a particular function or structure that allows it to be distinguished and can also be referred to as a subunit.
Protein structures
- Each polypeptide folds creating unique sections that are tailored for their particular function. If a protein unfolds, or denatures, and loses its conformation, it may no longer function. Protein structure, or conformation, is fairly complex and is organized into four categories (see Figure 11 below).
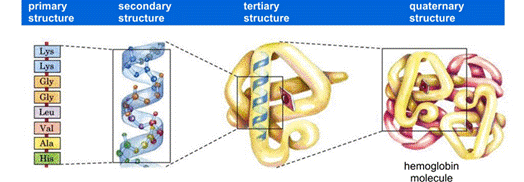
Figure 11: Categories of protein structures
- The primary structure of a protein is the sequence of amino acids in the polypeptide chain.
- By convention, the left end of a polypeptide primary structure (also known as amino acid sequence) is drawn as its N-terminus or N-terminal end, corresponding to the amine end with the unreacted amino group -NH2. The right end of the polypeptide is its C-terminus or C-terminal end, corresponding to the unreacted carboxyl group -COOH.
- As polypeptide chains fold up some areas of the chain form very regular folded patterns. These folded areas represent the secondary structure of the protein. Two patterns of folds are part of the secondary structure:
i) The alpha (α) helix: amino acids are arranged in a right-handed helix structure composed of 3.6 residues per turn.
ii) 74.2. Beta (β) sheets, are formed by beta-strands, that can be arranged parallel or anti-parallel to one another.
- Alpha helices and beta sheets are held together with hydrogen bonds that form between the atoms in the backbone of the polypeptide chain. When an alpha helix or beta sheet forms, the positively charged hydrogen atoms from the amino groups are attracted to the negatively charged oxygen atoms of the carboxyl groups. These weak electrical attractions act like molecular Velcro and hold the alpha helices and beta sheets in their shapes. See Figure 13:
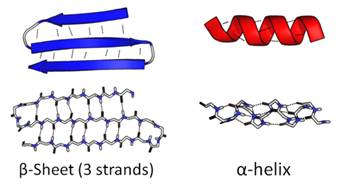
Figure 13: Secondary protein structures
- The tertiary structure is the final three-dimensional shape of the polypeptide chain. The final shape of any polypeptide chain is unique and will have specific areas that are necessary for the function of the protein.
- The 20 different amino acids found in proteins have 20 different R groups (also known as side chains). The structural biologist would know that different structures of the R groups give them different properties. As the polypeptide chain twists and folds upon itself, the R groups come into contact with each other. Depending on the structure of those two R groups, a bond may form between them (see Figure 14):
i) Covalent bonds: the amino acid cysteine has a sulfhydryl group (–SH) in its R group. When the R groups from two cysteines come near each other in a folded polypeptide chain, they can form a covalent bond called a disulfide bridge. "Di" means two, and disulfide bridges contain two sulphur atoms (–S–S–). Disulfide bridges are strong and are not lost when a protein denatures.
ii) Ionic bonds: some R groups can ionize so an ionic bond may form between them. These ionic bonds have variable strength.
iii) Hydrogen bonds: some R groups contain polar groups, meaning that there are slight differences in positive and negative charges in the atoms forming the chemical groups. When atoms with a slight positive charge come near atoms with a slight negative charge, hydrogen bonds form between them. These bonds are weak.
iv) Hydrophobic interactions: Some R groups are hydrophobic and can get pushed together in little pockets inside the folded polypeptide chain, forming a hydrophobic interaction. These bonds are weak.
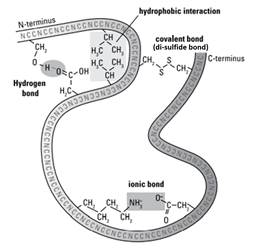
Figure 14: Types of bonds between the amino acids in a polypeptide chain.
- Some proteins consist of more than one polypeptide chain. These proteins have quaternary structure. The bonds that hold multiple polypeptide chains together to make a protein are the same types of bonds that hold together the tertiary structure of proteins. As proteins fold into complex quaternary structures, any changes to the amino acid sequence of a protein can have an impact on the way it folds and ultimately its biological function.
- One example of a protein with a quaternary structure is hemoglobin (as depicted in Figure 11 above). Hemoglobin consists of two pairs of different polypeptides, designated the α and β chains, which form a tetramer. These polypeptides are held together by hydrophobic interactions, hydrogen bonding, and ion pairs (salt bridges) between oppositely charged amino acid side chains.
- There are further more detailed points on proteins below, but it is necessary to address some basics of immunology and vaccinology first.
- There are two interconnected systems of response as part of the immune system: innate and adaptive. These two systems collaborate to protect the body against foreign invaders.
- Innate immunity includes soluble and cellular mechanisms that are evolutionarily primitive and aimed at preventing infection or quickly eliminating common invaders. Mediators of the innate immune response are activated upon recognition of general molecular patterns and capable of immediate response without additional conditioning or maturation. Adaptive immunity, in contrast, is stimulated by highly specific molecular sequences. This part of the system, which relies on B and T cells, must be activated by different foreign agents, matured, and amplified. Responses are therefore slower to develop and become effective but are much more specific.
- Adaptive immunity is characterized by specific recognition of invading foreign agents (such as viruses and bacteria) at a molecular level. Bacteria and viruses include many potential antigens, both on their surface and inside. Antigenic proteins of bacteria, viruses and other pathogens can generate a strong immune response. Such antigens may therefore be isolated and/or identified in order to assist in the development of vaccines.
- An antigen can be (and usually is) a large macromolecule. The small site on an antigen which is specifically recognized and bound by lymphocytes is called an epitope.
- The two main types of lymphocytes are commonly known as B cells and T cells. B cells express B cell receptors ("BCR"), membrane-bound proteins that bind to antigens.
- Each B cell expresses a BCR with a unique specificity to an antigen. B cells also produce antibodies, a soluble version of their BCR that bind the antigen, flagging them for destruction. Antibodies are synthesized only after antigenic stimulation of the relevant B cell (see Figure 1 below).
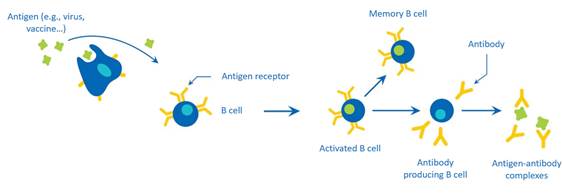
Figure 1: B-cell mediated immune response and production of antibodies upon encounter with an antigen.
- Both soluble antibodies and membrane-bound B Cell Receptors belong to the immunoglobulin family of proteins and consist of two identical heavy (H) chains and two identical light (L) chains kept together by intra- and inter-chain disulfide bonds (see Figure 2 below).
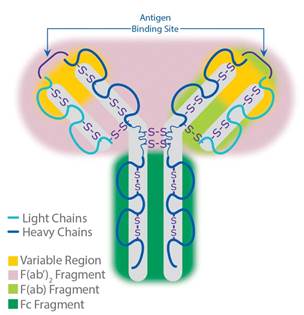
Figure 2: Antibody structure.
- Antibodies generated by activated B cells can protect the body against pathogens through several antibody-mediated effector functions. Collectively, these mechanisms result in the inhibition or destruction of pathogens, toxins, and cells in our body that have become harmful. Most viruses and some bacteria gain entry into a cell or tissue by binding specifically to one or more cell-surface proteins. Antibodies that bind such pathogenic proteins and block them from binding to cells or tissues are particularly potent effector molecules because they can prevent a pathogen from ever initiating an infection.
- Not all antibodies that bind to a pathogen are equally effective. Efficacy of antibody function depends on its site and strength of binding. When an antibody is capable of binding and affecting the biological processes of infection and pathogenesis, they can block the pathogen and neutralize its effect. They are referred to as neutralizing antibodies. This is in contrast to non-neutralizing antibodies which are capable of binding the pathogen in an antigen-specific manner but without inhibiting pathogen attachment to its cellular receptor. Collectively, neutralizing and non-neutralizing antibodies comprise the total antibody response generated. Neutralizing antibodies are important in the design of vaccines.
- A single antigen may have a number of epitopes, i.e., different sites separately recognized by the immune system.
- The epitopes of a protein can be either linear or conformational epitopes. A linear epitope is a certain sequence of amino acids. A conformational epitope is a set of amino acids, not necessarily a single linear sequence, present in a specific three-dimensional conformation when the protein is properly folded.
- A conformational epitope is therefore formed by the folding of the tertiary and/or the quaternary structure (see further below) so that you get remote parts of the same or different polypeptide chains that then come together to form that epitope. The epitope is the target for antibodies.
- There are different subsets of T cells - CD4 T cells, also known as helper T cells, and CD8 T cells also called cytotoxic T lymphocytes ("CTLs"). CTLs are important in clearing virus-infected cells, but do not directly prevent subsequent infections. Helper T cells play a central role in both humoral (mediated by antibodies) and cellular responses, through the particular molecules that they secrete. Two subtypes of helper T cells (Th1 and Th2) have been identified as being responsible for guiding adaptive responses towards either a cellular profile (Th1) or a humoral profile (Th2). Th1- polarised cells, which secrete interleukin-12 (IL-12) and interferon-γ (IFNγ) are responsible for the control of intracellular pathogens such as viruses and some bacteria. Th2-polarised cells are important in the defence against extracellular microorganisms, producing cytokines such as IL-4, IL-5 and IL-13 and promoting eosinophilia, which can be damaging as part of the inflammatory process of allergic disease.
- The RSV Vaccinologist would have been aware that neutralizing antibodies are important in the design of vaccines. The binding sites and conformational requirements of these antibodies are therefore of interest to the vaccine designer. The production of neutralizing antibodies in response to a vaccine candidate would have been studied by the RSV Vaccinologist as a potential correlate of protection when undertaking clinical testing of vaccines.
Virus Structure and Function
- Viruses are not cells. Rather, they are microscopic particles of nucleic acid and protein that are incapable of independent replication. They attach themselves to cells, enter, and hijack the host cell materials and protein production machinery to produce more viral particles. Without a cellular host, viruses are inert.
- Viruses have different shapes and patterns of multiplication within cells, and the ones that attack humans cause many significant diseases. The simplest viruses have just two components: a nucleic acid core and protein capsid. The nucleic acid core, which may be DNA or RNA, contains the instructions for taking over cells and making more viral particles. The nucleic acid is surrounded by the capsid, a protective protein coat. Some viruses have an outer membrane layer called an envelope, and these are called enveloped viruses. The envelope of a virus is similar to the plasma membrane of a cell with the addition of certain viral proteins. The viral proteins that protrude from the envelope or the surface of the capsid help the virus attach and enter host cells.
- The viral genome consists of one of four types of nucleic acid: double-stranded DNA (dsDNA), single-stranded DNA (ssDNA), double-stranded RNA (dsRNA) and single-stranded RNA (ssRNA).
- Single-stranded RNA (ssRNA) can be divided into two groups: positive-sense ssRNA viruses and negative-sense ssRNA viruses. Positive-sense ssRNA viruses are those in which RNA molecules can be read directly for the synthesis of viral proteins. On the other hand, negative-sense ssRNA viruses are those in which their RNA molecules cannot be read directly to create proteins. Instead, their negative sense RNA molecules must first be used to make complementary, or "mirror image" positive-sense RNA molecules. Then, these new complementary RNA molecules can be translated to make viral proteins.
- Viruses attach to cells when viral proteins successfully bind to receptors on the host cell. If the viral protein has the right shape, it will tuck into the corresponding, complementary shape on the host cell receptor. Viral attachment may be thought of as a virus having the right key to fit into the lock on the host cell. After the virus is attached, it may enter the cell by creating a hole in a cell membrane, slip in by fusing its envelope with the membrane of the host cell, or induce the cell to actively import it. The ability of a virus to infect a host cell depends on a match between attachment proteins on the surface of the virus and receptors on the surface of the host cell.
- Viral infection of a cell begins with the attachment of viral proteins to receptors on the host cell. After the virus binds to the host cell, it crosses the plasma membrane of the host.
- In order to reproduce, viruses need to copy their genetic material and make more viral proteins. In human cells, production of its nucleic acids (DNA and RNA) occurs in the nucleus, while synthesis of proteins occurs in the cytoplasm. Viruses use the machinery of the host cells they have infected to produce their own genetic material and proteins to produce new virus particles. Viruses replicate either in the nucleus or the cytoplasm with most RNA viruses producing their RNA genomes and mRNA molecules in the cytoplasm of the host cell using the virally encoded polymerase.
- The goal of all vaccines against infectious disease is to expose the individual to the antigen(s) of a pathogen in a safe way, in order to elicit the development of specific and long-lived adaptive responses, so that the vaccinated individual will be protected in the future when they encounter the real pathogen.
- One mechanism by which vaccines work is by taking advantage of antigen recognition and the antibody response. A vaccine contains the antigens of a pathogen that causes disease. By way of example:
i) the MMR vaccine is a vaccine against measles, mumps, and rubella. It contains viruses that have been weakened through multiple replication passages in cells. The viruses are structurally intact and therefore include all the specific antigens, but they do not normally cause disease in healthy individuals. When a person is vaccinated with the MMR vaccine, the immune system responds by stimulating antibody-producing cells that are capable of making specific antibodies; and
ii) the hepatitis B vaccine contains only the viral surface antigen (not the whole virus). When a person is vaccinated against hepatitis B, the immune system responds by stimulating antibody-producing cells that are capable of making antibodies specific for the viral surface antigen.
- Immunologic memory harboured by residual B and T lymphocytes is the foundation for vaccination, which uses crippled or killed pathogens or pieces of them as a safe way to "educate" the immune system to prepare it for later attacks by disease-causing pathogens. Memory cells then save the strategy used, but not the pathogen (or vaccine), for later reference during repeat encounters with the same infectious agent.
- When vaccination works, the continued exposure of activated T and B cells to the antigen leads to the selection (from an accumulation of single point mutations) of the T cells expressing the highest affinity T cell receptors and the "fine-tuning" of the B cell receptors. This "experiential learning process" gradually increases the affinities over time and can produce extremely specific and long-lived memory cells, capable of protecting the host from the pathogen for many decades. As a result, if the body comes into contact with the pathogen in the future, the body is prepared to fight it.
- At the Priority Date (and still today), the RSV vaccinologist understood that the first crucial step in the path to a new vaccine is to define specific immunologic targets. These targets are generally the antigenic determinants or conformational epitopes of a pathogen and represent the specific immunologic goals or markers that scientists believe will result in protection (immunity) from infection or disease upon natural encounter with that pathogen.
- As at the priority date (and still today), vaccines for humans can be classified in several types:
i) Live Attenuated Vaccines: the viruses or bacteria are weakened so that they lose their ability to cause significant disease in healthy subjects but retain their capacity for slow and transient growth which allows the immune system an exposure to the real pathogen so that it produces antibodies and T cells. Many such vaccines had been approved for human use.
ii) Inactivated or "Killed" Vaccines: by treatment with heat or chemicals. This kills the pathogen, making it incapable of replication while preserving some portion of the pathogen's antigens, allowing it to induce an immune response to at least some of the antigens. Many such vaccines had been approved for human use.
iii) (Purified or Recombinant) Subunit Vaccines: use only specific molecules from the pathogen. These can be made by either growing the pathogen and purifying part of it after disrupting its structure or by using recombinant protein technology. The antigen product(s) must retain the conformation required to elicit sufficient immune responses. Increasing numbers of such vaccines, including those comprising recombinant proteins, have been approved for human use.
iv) Recombinant Vector Vaccines: individual genes encoding viral antigens can be introduced into "non-harmful" viruses or bacteria that are used as live carriers, producing the antigen, or as an inactivated carrier for delivery of DNA encoding the antigen. At the Priority Date, no such vaccines had been approved for human use, but the approach was subject to active research.
v) mRNA and DNA Vaccines: synthetic mRNA or plasmid DNA encoding antigenic proteins are injected directly into the recipient. The host cells of the recipient then produce the antigen protein in vivo. At the Priority Date, no such vaccines had been approved for human use, but the approach was subject to active research.
History and Classification of RSV
- RSV was first isolated from chimpanzees in 1956 and subsequently recovered from infants with severe lower respiratory tract disease.
- At the priority date, RSV was classified in the family Paramyxoviridae. The family Paramyxoviridae (containing the various paramyxoviruses) was classified into two subfamilies: the Paramyxovirinae and the Pneumovirinae. The Paramyxovirinae subfamily included, among others, mumps virus, measles virus, Sendai virus, Newcastle disease virus ("NDV"), parainfluenza virus 5 ("PIV5"; formerly known as simian virus 5 ("SV5")), human parainfluenza viruses ("hPIV") 1–4, and the deadly Nipah and Hendra viruses. RSV was a member of the subfamily Pneumovirinae, genus Pneumovirus. The Pneumovirinae subfamily also included, among others, bovine RSV ("BRSV"), pneumonia virus of mice ("PVM") and human metapneumovirus ("hMPV").
Epidemiology
- RSV causes annual epidemics during the winter in temperate climates with the annual epidemic peaking around December in the UK.
- Historically, RSV was considered a pathogen of early childhood as more than 90% of infants are infected with RSV before they turn 2 years old. Although normally resulting in upper respiratory tract infection with mild-to-moderate symptoms, in a subset of infants, disease progresses to a more severe infection of the lower respiratory tract (bronchiolitis), and pneumonia requiring hospitalization.
- However, RSV also causes a substantial disease burden among older adults who live in the community or in long-term care facilities.
- By the priority date, it had been known for many years that RSV is only weakly immunogenic and could repeatedly reinfect immune individuals.
- There are two subgroups of HRSV; HRSV-A and HRSV-B. The main difference between the two subgroups is in the attachment protein (discussed in more detail below). Both of these subgroups cause disease.
Morbidity and Mortality
- Hospitalization rates of infants infected with RSV vary with the setting. Hospitalization rates for children in lower socioeconomic groups tends to be significantly higher. Overall, the rate of hospitalization for RSV disease in developed countries was approximately 1 to 20 per 1,000 infants younger than 1 years old, with 9% of patients requiring mechanical ventilation.
- By the priority date, mortality due to RSV infection was uncommon in children in developed countries. There were no exact determinations of the overall death rate, but the estimates dropped over the course of the second half of the 20th century. A survey from the mid-1970s estimated the fatality rate in the UK at 0.5% to 2.5% of hospitalized children with RSV infection. However, as a result of more effective modern intensive care, the estimates as at the priority date were as low as 0.3% of hospitalized children.
- RSV also causes significant morbidity and mortality in the elderly. RSV had been shown to develop annually in 3 to 7% of healthy elderly patients and in 4 to 10% of high-risk adults. RSV infection accounted for 10.6% of hospitalizations for pneumonia, 11.4% for chronic obstructive pulmonary disease, 5.4% for congestive heart failure, and 7.2% for asthma. The mortality rate associated with RSV pneumonia in adults with profound immunosuppression due to leukemia or hematopoietic stem cell transplant was shown to be as high as 80 to 100%.
Pathogenesis of RSV
- RSV is spread by respiratory secretions and is one of the most infectious human viruses. Contamination by large droplets or of fomites (objects or materials which are likely to carry infectious viral particles, such as toys, clothes, utensils, and furniture) are thought to be the major modes of spread. Contact with infected individuals or contact of contaminated hands to nasal or conjunctival mucosa is required for the virus to spread. The incubation time from infection to onset of illness normally is 4-5 days.
- The nasopharynx is the initial site of virus replication. In susceptible groups, the virus is able to spread rapidly to the lower respiratory tract which results in symptoms 1 - 3 days after the onset of rhinorrhea (a runny nose). Spread of the virus from upper to the lower respiratory tract likely involves aspiration of virus-containing secretions.
Immune Responses in RSV Infection
- At the priority date, it was known that the adaptive immune response plays the primary role in host defense during RSV infection and resistance to reinfection.
- At the priority date, it was known that protection against reinfection by RSV was conferred by:
i) CTLs, which likely contribute to short-term protection, such as against reinfection during the same epidemic;
ii) local secretory IgA antibodies which appear to play a major role in short-term protection and, especially following multiple infections, in long-term protection; and
iii) serum antibodies, which confer durable protection that is often not complete, especially in the upper respiratory tract and which may be passed transplacentally, providing the neonate with maternal antibodies that may provide some protection against infection during the first few months of life.
- The RSV genome is 15.2 kb of non-segmented single-stranded negative-sense RNA encoding 11 viral proteins. The viral envelope of RSV contains three transmembrane glycoproteins: attachment glycoprotein ("G"), fusion protein ("F"), and small hydrophobic protein ("SH"). Matrix proteins ("M") are present on the inner side of the viral envelope. Viral RNA is tightly encapsidated by the nucleoprotein ("N") while the large protein ("L", the viral polymerase), phosphoprotein ("P"), and M2-1 protein mediate viral RNA transcription. M2-2 protein regulates viral RNA synthesis
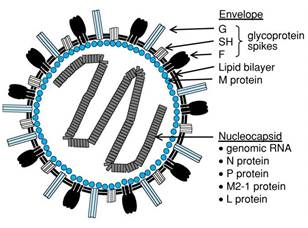
Figure 4: The structure of respiratory syncytial virus (RSV).
- The genes encoding the viral proteins are represented linearly according to their position in the viral genome. A linear drawing of the HRSV genome is shown as Figure 3, below.
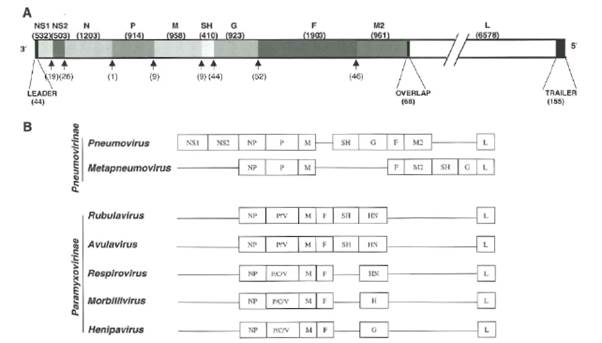
Figure 3: Scheme of the HRSV genome (A) The different genes of the HRSV A2 strain are shown at scale, except for the L gene that is split into two fragments. The nucleotide lengths of gene and intergenic regions are shown between parentheses above and below the genome diagram, respectively. Also shown are the leader, trailer and overlapping segment between M2 and L genes. (B) Comparison of the gene order in different genus of the Pneumovirinae and Paramyxovirinae subfamilies.
- As indicated in Fig 3(B), the paramyxoviruses share similar structural components with one another. They each have a lipid envelope into which glycoproteins are inserted. Glycoproteins are proteins to which carbohydrate chains are bonded at certain sites.
- The SH glycoprotein is only found in some paramyxoviruses. Unlike other paramyxovirus F proteins (which only have one furin cleavage site in the F protein), HRSV and BRSV have two furin cleavage sites in their F protein.
- RSV particles (virions) vary in shape and size. When observed by electron microscopy, two types of viral particles have been identified: (i) round- or kidney-shaped particles ranging from 150 to 250 nm wide and (ii) filaments up to 10μm in length. Both types of particles are infectious.
- RSV attaches to the host cell when viral proteins interact with target molecules (receptors). Attachment is mediated by RSV F and G glycoproteins anchored into the viral envelope through transmembrane regions with RSV F naturally forming trimers. Following attachment, viral entry into the host cell occurs upon triggering of the F protein to initiate fusion of the viral envelope and host cell plasma membrane. After the internalization of the viral nucleocapsids into the cell cytoplasm, sequential transcription of the viral genome is activated to generate a set of mRNAs that instruct translation of the corresponding gene products by the cell ribosomes. Eventually, the different RSV gene products accumulate at the cell membrane where they are assembled into progeny virus particles that are released from the infected cell by budding.
- Virus entry by enveloped viruses has been a topic of intensive investigation. Two steps are well differentiated in this process:
i) binding of the virus to certain cell surface components;
ii) fusion of the virus and cell membranes at the cell surface.
- The RSV G and F glycoproteins mediate these two steps, respectively. However, the RSV Vaccinologist would have been aware that G enhances the process, but it is not required for infection of certain cell types in tissue culture. The RSV Vaccinologist would have also known that G and F are the major glycoproteins on the surface of the virion and have important roles in entry. The G glycoprotein functions primarily as an attachment protein that binds virions to target cells by interacting with one or more host cell surface molecules. The F glycoprotein can also facilitate attachment, although to a lesser extent than G, but its primary function is to mediate fusion of the viral and host cell membranes.
- Unlike most other paramyxoviruses which have a requirement for the attachment (HN) protein in mediating fusion, the RSV F protein can (at least in vitro) mediate fusion in the absence of the G protein.
- A theoretical model of how the process of fusion of an HRSV particle to a target cell membrane was thought to occur is shown in Cane at page 325.
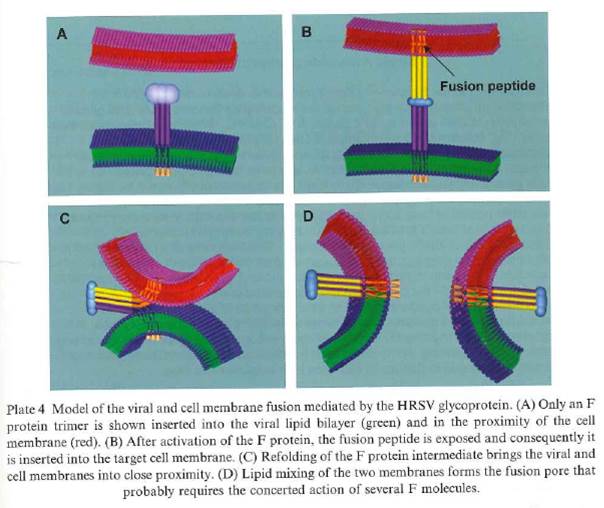
Figure 5: Theoretical model of the viral and cell membrane fusion mediated by the HRSV F protein. The viral lipid envelope is shown in green and the target cell membrane is shown in red. The purple and yellow stalks represent the heptad repeat (HR) regions A and B, with the fusion peptide at the N'-end of HRA.
- Once the HRSV particle has attached to the cell, the fusion peptide is exposed and inserts into the target cell membrane. This is the step shown in the image labelled B above. The viral envelope and cell membranes are then pulled together, allowing mixing of their lipids and formation of the fusion pore.
- The process of fusion enables the viral RNA to enter the cells where new viral proteins are synthesised and the RNA is replicated. The replication cycle of HRSV is shown in Figure 6 below:
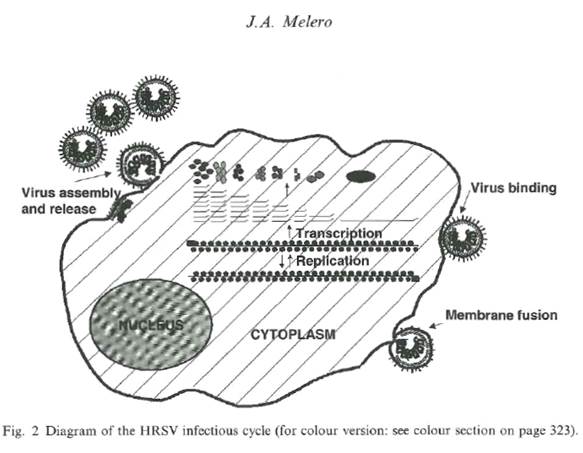
Figure 6: Diagram of the HRSV infectious cycle showing virus binding and fusion with a host cell, intracellular processing within the cell, virus assembly at the cell membrane and budding of virus progeny from the cell surface.
F Glycoprotein
- The F protein is a class I glycoprotein synthesized in the cytoplasm of a virus-infected cell as an inactive precursor of 574 amino acids (F0). In contrast to other paramyxovirus F proteins that are cleaved only once, the precursor of RSV is subsequently cleaved at two sites by a cellular furin-like protease to yield two domains: a larger carboxy-terminal domain F1 and a smaller amino-terminal domain F2. The F2 and F1 domains are covalently linked to form a heterodimer through two disulfide bonds.
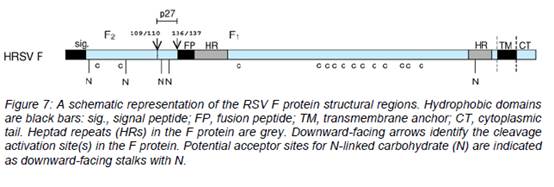
- A schematic representation of the RSV F protein structural regions is shown in Figure 7 below.
- The furin-like proteases of the trans-Golgi cleave F0 during its maturation after residues 109 (site I) and 136 (site II). Site II is equivalent to the single cleavage site found in other paramyxoviruses. The 27 amino acid peptide that is released after maturation of the F0 precursor is known as peptide 27, p27 or pep27. Cleavage is not always complete and sometimes uncleaved F0 molecules and partially processed intermediates are formed when cleavage occurs at only one site.
- The mature active form of the F protein present on the surface of the virus and infected cells comprises the F2 and F1 domains linked by disulphide bonds (a covalent bond that forms between two cysteine residues).
- There are three main hydrophobic regions on the HRSV F protein shown as black sections in Figure 7 above: one at the N-terminus (left) of F0 which has a role as a signal peptide (sig.) during synthesis, one near the C-terminus (right) of F0 which is the transmembrane domain (TM), and one towards the N-terminus of F1 (at the C-terminus of pep27 (p27) and cleavage site 136/137) which is called the fusion peptide (FP). The fusion peptide region is a hydrophobic glycine-rich segment that inserts into the target cellular membrane during the fusion process, The transmembrane region keeps the F protein anchored in the lipid envelope of the virus particles. The fusion peptide is the region of the F protein which mediates fusion.
- Adjacent to both the fusion peptide and transmembrane regions are two regions with heptad repeat sequences containing a motif suggestive of coiled-coil structures. These regions are denoted generally in the literature as HRA and HRB, (sometimes HR-N and HR-C for the N-terminal and C-terminal HR regions of the F1 domain), respectively, and are separated by an intervening region of about 250 amino acids.
- A coiled-coil is a structural motif in proteins in which α-helices (i.e., coils) are coiled (twisted) together like the strands of a rope. In the F protein, HRB forms a coiled-coil in the formation of a trimer.
- The mature F protein is transported to the cell membrane where its transmembrane domain is anchored in the cell membrane and the extracellular domain of the F protein extends into the surrounding medium. In this way, the F protein is presented on the surface of the infected cell. The mature F protein present on the surface of the virus and infected cells consists of a homotrimer of three non-covalently associated units of disulfide-linked F1–F2 with a resulting molecular weight of approximately 210 kDa; however, the 3-dimensional structure of the HRSV F protein (or the BRSV F protein) had not been solved, although models had been generated.
Prefusion and postfusion conformations of the F glycoprotein
- By the Priority Date, electron microscopy studies by a group led by Dr Melero from Spain in collaboration with Professor John Skehel in the UK, demonstrated two apparent forms (or conformations) of HRSV F protein rods and proposed that these may represent the different conformations that the F protein adopts before and after activation for its role in membrane fusion. These rods were described as cone-shaped rods and lollipops. It was also observed that rosettes arose upon aggregation of each protein form, and involvement of the fusion peptide in the F protein was hypothesized to play a role in this. The resulting Calder paper (2000) reported that two distinct types of RSV F protein rods were identified by electron microscopy, which "may represent different structures that the molecule may adopt before and after activation for its role in membrane fusion". It was known at the priority date that the HRSV F protein had two conformations.
- I set out the preceding paragraph in full because it was agreed to be CGK, but Pfizer and Dr Johnson said the CGK extended beyond that. This is a major area of dispute as to the CGK, which I address below.
- By the priority date, the F and G proteins had been identified as the major protective antigens of HRSV. Studies in rodents and calves demonstrated that the F and G proteins are important in generating protective immunity. Although infection with HRSV induces antibodies against a variety of viral proteins, only those specific for the F and G proteins include neutralising antibodies.
- RSV can be subdivided into two groups, A and B. The subgroups could be distinguished antigenically with polyclonal sera and monoclonal antibodies (mAbs). Sequence analysis of representatives of the two subgroups showed they share 81% nucleotide identity. It was known that the F protein of the two subgroups is relatively stable antigenically. The F protein is well conserved between HRSV-A and HRSV-B (with 89% amino acid identity between the subgroups), whereas there is only 53% amino acid identity between the G proteins of HRSV-A and HRSV-B strains.
Antiviral Compounds
- Ribavirin (a nucleoside analogue) was approved for use in the US for treatment of RSV infection but did not perform well in the clinic.
Anti-inflammatory Agents
- Attempts had been made to develop anti-inflammatory treatments to reduce respiratory tract disease.
Bronchodilators
- Drugs used to treat reversible airway smooth muscle constriction in asthma were also tested in infants with more severe RSV disease.
Passive immunoprophylaxis
- In the absence of a vaccine at the priority date, passive immunoprophylactic treatment with polyclonal or monoclonal antibodies had been shown to be an effective approach to reducing RSV disease severity.
- Approaches included monthly intravenous infusions of human immunoglobulin ("RSV-IVIG") (prepared from sera of donors pre-screened for high RSV-neutralizing activity). This product was later licensed in 1996 for infants at high risk of serious RSV disease and given the brand name RespiGam. However, RespiGam had major drawbacks, including the large volume of infusion and the inconvenience of repeated administration. In addition, it had the potential to interfere with other childhood vaccinations due to the presence of antibodies specific for other pathogens.
- A mAb, palivizumab (branded Synagis), was later licensed in 1998. Palivizumab was based on a murine mAb specific for the F protein and successfully neutralizes both RSV subgroups (A and B). The advantage over RespiGam was that Synagis was 50- to 100-fold more effective on a weight basis in neutralization and therefore the total amount of immunoglobulin which needed to be administered could be greatly reduced. Synagis is administered by an intramuscular injection on a monthly basis. However, because of the expense associated with such a prophylactic approach, its use was (and still is) limited to premature babies or infants with congenital cardiopulmonary conditions.
- Given that the evidence at the Priority Date indicated that it might not be possible to provide complete protection against infection, the ultimate aim in the field was to develop a vaccine that provided sufficient protection to prevent serious lower respiratory tract disease leading to hospitalization and to decrease the frequency of complications. There was a worldwide need for at least two, likely different, vaccines: (1) a paediatric vaccine for the RSV-naive and (2) a vaccine for RSV-experienced individuals at increased risk of serious disease, including the elderly and individuals with chronic pulmonary or cardiac disease.
- Multiple approaches to HRSV vaccines had been evaluated in the clinic by the priority date. All of these types of vaccines induce an immune response by generating neutralising antibodies. Live-attenuated, vectored and DNA vaccines also have the advantage of inducing CTLs. Inactivated RSV was generally not considered as a viable vaccine candidate due to the adverse outcome of the formalin-inactivated RSV vaccine trial (discussed below).
Formalin-inactivated RSV vaccine
- The development of an RSV vaccine was severely affected by the issue of enhanced disease associated with a formalin-inactivated RSV vaccine ("FI-RSV"). Formalin is an aqueous solution of formaldehyde (an organic compound) that kills microorganisms by cross-linking amino acids and so rendering proteins non-functional. Formalin has been used to make inactivated vaccines against other viruses. FI-RSV contained RSV that was grown in vitro, inactivated with formalin, concentrated, and mixed with aluminium hydroxide adjuvant. FI-RSV was administered intramuscularly to infants and young children in multi-centre trials in the 1960s.
- The vaccine was poorly protective and had the paradoxical effect in RSV-naive patients of increasing the severity of RSV disease during subsequent natural infection. In one vaccination centre, 80% of vaccines required hospitalization compared to only 5% in the control group and two of the FI-RSV vaccinated infants died after being naturally infected with HRSV.
- The RSV Vaccinologist would have appreciated that this disease enhancement due to FI-RSV was not fully understood at the Priority Date, but one theory for the low level of neutralising antibodies induced by the FI-RSV vaccine was that the epitopes on the F protein involved in inducing neutralising and fusion-inhibiting antibodies had been modified in some way by the formalin inactivation.
- This history of HRSV vaccine-enhanced lung pathology has been a major challenge to RSV vaccine research and delayed progress on RSV vaccine development for a number of years. Safety and efficacy concerns varied depending on the target population of the vaccine and as a result, different vaccination strategies were employed and preferred for different target populations. Vaccine-induced enhanced disease was considered a major concern for RSV-naive infants. In contrast, target populations of the elderly adult and pregnant women were not considered at risk of vaccine-induced enhanced disease due to pre-existing memory immune responses.
- Following these encountered disease enhancement issues, inactivated RSV was generally not considered as a viable vaccine candidate at the priority date. A prominent school of thought was that the risk of enhanced lung pathology was caused by epitopes on the native F protein which had been denatured or modified during the process of inactivation with formalin. It was therefore thought that the vaccine-enhanced pathology could be minimised by seeking to present the virus or its proteins in the form in which they are naturally expressed. This was thought to be safest because using the virus in its native form should produce an immune response that is similar to that induced by natural infection. This point is relevant to a dispute which arose over the use of the term 'native'.
Live-attenuated vaccines
- One focus of RSV vaccine development at the priority date was designing a live-attenuated vaccine for intranasal administration in paediatric populations. An intranasal live-attenuated RSV vaccine mimicked natural infection since a limited amount of viral replication occurred and was presumed to induce the same local and systemic immunity.
- Studies in experimental animals confirmed that intranasal infection with attenuated strains of RSV was highly immunogenic and protective. Clinical studies showed that an experimental live-attenuated RSV vaccine readily infected and immunized 1- to 2-month-old infants (despite the presence of maternal antibodies), and there was no association with enhanced disease.
- It was known that a number of live-attenuated RSV vaccine candidates had been developed at the priority date. The most promising set of biologically derived live vaccine candidates were developed by serial passage of RSV at increasingly suboptimal temperatures (termed cold passage ("cp")) followed by chemical mutagenesis and screening in order to identify mutants which were temperature-sensitive ("ts") mutants. This strategy was designed to introduce mutations that would attenuate the virus for growth under normal physiological conditions. These were being developed for intranasal vaccination. However, they were found to be either under-attenuated or over-attenuated.
- One derivative, cpts248/404, seemed to be a promising vaccine candidate, but was associated with upper respiratory symptoms and a restricted immune response in infants 6 months of age.
- Reverse genetics was an alternative approach to conventional biological approaches for producing attenuated derivatives. This approach became popular from the 1990s onwards and involved introducing mutations in the virus genome, or deleting genes not essential for replication. The virulence of the genetically modified viruses was then analysed in an animal model to identify if it had been attenuated. Mutations identified in the biologically-derived cold-adapted and ts mutants were introduced into recombinant wild-type HRSV to determine their role in the attenuating phenotype. Examples of non-essential genes that were deleted to produce attenuated HRSV were NS1, NS2, SH, G, and M2-2. Some of these gene deletions were combined with mutations shown to play a role in temperature restricted replication.
- Another strategy was based on BRSV. At the priority date, studies were underway to replace additional BRSV genes with their RSV counterparts. One approach replaced the F and G genes of BRSV with those of HRSV, but the chimeric virus was still over-attenuated. The expression of the protective antigens in genetically engineered RSV was increased by moving the G and F genes from their natural positions as the seventh and eighth genes in the gene order to promoter-proximal positions.
- In another approach, B/HPIV3-F and B/HPIV3-G chimeric viruses were developed, which were based on bovine parainfluenza virus 3 (bPIV3) in which the F and HN genes were replaced by the F and HN genes from human PIV3 and the HRSV F gene G gene was inserted. These chimeric virus were highly immunogenic in non-human primates.
- A number of approaches to improving the immunogenicity of attenuated RSV vaccines, such as incorporation of immunomodulatory genes (e.g. interleukin (IL)-2 or IFNy) had been described by the priority date.
Protein subunit vaccines
- At the priority date protein subunit vaccines were the subject of clinical trials. Studies had shown that protein subunit vaccines could generate neutralizing antibody titres and protective responses in animals. By the priority date, protein subunit vaccines had been evaluated in RSV-experienced individuals and found to be safe and immunogenic. Both purified and recombinant subunit vaccine approaches were being explored.
- As already mentioned, at the priority date the G and F glycoproteins represented the major targets for the humoral immune response against RSV as they induce strong neutralizing antibody responses, with F generating a greater magnitude of neutralizing antibodies than G. Antibodies are the major correlate of protection in many viral infections and with most licensed vaccines. G was included in some subunit vaccines since the greatest variation between RSV A and B types was found in the G glycoprotein. However, inclusion of G in the vaccine often induced type 2 T cell responses that increased disease severity on challenge. While these disease-enhancing immune responses could be partially modulated by using of Th1-biasing adjuvants, safety concerns on the inclusion of G persisted. Thus, as RSV F generated both antibody and CTL responses, including potent neutralizing antibodies, and was highly conserved across all RSV types, RSV F was the most attractive target for protein subunit vaccines at the Priority Date.
- One such vaccine approach developed by Wyeth in the 1990s before there had been any recognition of the differing conformations of the F protein consisted of using purified F protein (PFP) which was isolated from RSV-infected cell culture and formulated with aluminium hydroxide adjuvant. Several PFP vaccines were developed and designated PFP-1, PFP-2, and PFP-3. Studies had shown that mice or cotton rats immunized with purified F protein generated serum neutralizing antibodies and CD8 cytotoxic T cell responses that protected them against live virus challenge. Additionally, in chimpanzees previously immunized with RSV, purified F protein vaccination produced a greater than 4-fold increase in neutralizing antibody titres. The vaccines had been evaluated extensively in adults, children with and without underlying disease, and in the elderly. The vaccines were immunogenic and well tolerated with minimal acute reactions. Enhanced disease was also not observed.
- A Phase I study of PFP-2 was conducted in pregnant women during their third trimester with the aim of achieving higher titres of RSV-neutralizing maternal antibodies in serum and milk which would result in increased resistance to RSV infection and disease in the newborn infant. The vaccine was well-tolerated and induced a fourfold or greater increase in antibody titres in almost all of newborns of vaccinated mothers compared to none of neonates of placebo-immunized controls. However, only a modest increase in neutralizing antibody titres was observed.
- The RSV Vaccinologist would also have known that another Phase I study of PFP-2 was conducted in children with cystic fibrosis. Although protection against RSV infection was not observed, a significant reduction in mean number of lower respiratory tract illnesses, antibiotic courses and days ill occurred among RSV-infected vaccinees. The third generation PFP-3 vaccine was tested in a larger Phase III multicentre study performed in children with cystic fibrosis. While the vaccine was well-tolerated and induced a fourfold rise in neutralizing antibody titres, it was not associated with significant protection.
- The RSV Vaccinologist would also have been aware that a vaccine consisting of co-purified F, G and M proteins from RSV A had been tested in healthy adults with either aluminium or polydicarboxylatophenoxyphosphazene (PCPP) as an adjuvant. The vaccine was well tolerated, and 4-fold or greater rise in neutralizing antibody titres was detected in majority of vaccinees. Neutralizing antibody titres waned after one year but could be boosted by revaccination.
- With RSV F and G glycoproteins expressed on the surface of viral particles and infected cells and as the primary targets of most neutralizing antibodies induced by RSV, a recombinant chimeric FG fusion protein vaccine was evaluated by multiple groups. One laboratory consistently reported induction of neutralizing antibody responses that protected the lower respiratory tract, but not the upper, against RSV infection with no increase in disease severity. In contrast, other investigators demonstrated induction of non-neutralizing antibodies, type 2 T cell responses, and enhanced disease when the FG subunit vaccine was formulated with alum-based adjuvants or if RSV exposure was more than 1 month after immunization. However, they also showed that delivery of FG with a Th1-inducing adjuvant such as QS-21 or CpG oligodeoxynucleotide, in the form of an immune-stimulating complex, or by a viral vector induced protective neutralizing antibody responses without increasing disease severity. One chimeric FG antigen was evaluated in unreported Phase I trials. However, at the Priority Date, the RSV Vaccinologist would have been aware that the development of FG chimeric antigens had ceased.
- The RSV Vaccinologist would also have known that another protein vaccine, BBG2Na, had also been reported to be safe and highly immunogenic in adults and the elderly. BBG2Na consisted of a recombinant bacterially expressed fragment of the G protein that contains the conserved central domain (amino acid 130-330) fused to the albumin-binding region of the streptococcal G protein. However, there were reports of unexpected side effects. BBG2Na was no longer in clinical development by the Priority Date.
- By the priority date, virus-like particles ("VLPs") were being investigated as an alternative platform for RSV subunit vaccines. VLPs are non-replicating, non-infectious particles derived from virus-encoded proteins. For example, the hepatitis B surface antigen (HBSAg), which can form VLPs, had been engineered to contain an RSV-encoded CTL epitope. These VLPs and nanoparticles derived from bacterially derived RSV nucleocapsid (N) proteins conferred a moderate degree of protection against RSV challenge in rodent models.
- Studies had demonstrated that intranasal vaccination of mice with genetically detoxified cholera toxin administered with F protein or a peptide corresponding to amino acids 174 to 187 of the G protein induced protection against RSV. Similarly, intranasal vaccination with purified RSV surface glycoproteins formulated as immunostimulating complexes (ISCOMs) protected mice against RSV, as did intramuscular vaccination.
- Studies using a 21-mer G peptide (amino acids 170-190) expressed as a fusion with the alfalfa mosaic virus ("AMV") coat protein, such that the recombinant AMV particles expressed the peptide on their surface, and a chimeric protein consisting of amino acids 125-225 of the G protein linked to a CTL epitope of M2-2 formulated with aluminium hydroxide has also been described. As had a recombinant protein obtained by fusing the N-terminus of the chimeric CTL epitope F/M2(81-95) from HRSV to a carrier protein DsbA (disulphide bond isomerase).
Other RSV vaccine strategies
- Other RSV vaccine strategies were being investigated at the priority date including:
i) Synthetic peptides: RSV F and G have independently been used in rodent models for development of peptide-based vaccines.
ii) DNA vaccines: A DNA vaccine is comprised of a plasmid, a DNA sequence encoding genes of interest and any genetic regulatory elements required for the expression of the encoded genes, and possibly additional molecules to facilitate passage of the DNA plasmid across the host cell membrane and then the nuclear membrane of the cell.
iii) Vector-based vaccines: Vectored vaccines are engineered to express heterologous genes encoding the antigens of interest. The vector may also be modified so that it is unable to replicate and produce progeny virions but still able to express encoded proteins. Alternatively, live vectors (that use another microbe, such as another virus, as a vehicle) may be used that undergo limited replication so that no disease is produced, and this is accomplished by selecting a vector that is not a natural pathogen of the target population or by attenuating the vector by molecular engineering. For example, vaccinia virus, which was used as a live vaccine against smallpox, was engineered to express different HRSV proteins. Studies to evaluate the efficacy of vaccinia virus-vectored HRSV vaccines were undertaken in small animal models in the early 1980s. Replication-defective adenovirus vectors and the bacterial vector Staphylococcus carnosus had been used to express RSV F or RSV G. Replication-competent vector systems that had been utilized included modified vaccinia Ankara (MVA), paramyxoviruses (including PIV-1, PIV-2, PIV3, Sendai virus, and New Castle Disease virus), vesicular stomatitis virus, and the alphavirus Semliki Forest virus. These vectors were engineered to express RSV F or RSV G. Some vector systems were not sufficiently immunogenic, while others induced enough B and T cell immunity to protect against RSV infection without increasing disease. Virus-vectored vaccines, such as recombinant vaccinia virus (rVV), MVA and replication-competent adenovirus expressing RSV F and/or G proteins, and DNA vaccines were found to be immunogenic but did not confer consistent protection in chimpanzee.
Models of RSV infection
- At the priority date, there were a number of animal models available for RSV infection. The most commonly used animal models were as follows.
The mouse model
- The mouse model had the advantage of both being a small animal model (making housing easier) and being easily obtainable from multiple commercial sources. In addition, inbred mouse strains, providing matched genetic backgrounds, reduced variation between subjects and enabled improved statistical comparison relative to work performed in outbred (and so genetically mismatched) subjects.
- Although differences in virology existed between RSV-infected humans and mice, many aspects of RSV-induced immunity were recapitulated in the mouse model, most importantly induction of FI-RSV vaccine-enhanced disease and of cytotoxic CD8 T cell responses highly effective in eliminating RSV infection. Notably, development of the RSV mouse model coincided with an explosion of discovery and development in the field of immunology, providing new tools such as monoclonal antibodies and new technologies such as PCR and flow cytometry for detailed characterization of both human and mouse immune responses. Use of genetically modified transgenic mice also allowed fine dissection of multiple aspects of immunopathogenesis of RSV.
- RSV was not a natural rodent pathogen and required about 1,000-fold more viral particles than do humans for infection to be established, defining the mouse as a semi-permissive model in which more aggressive infection conditions (e.g., more virus particles or larger inoculum volume) are needed to achieve infection. It was considered this may be due in part to the fact that RSV infected different subsets of respiratory epithelial cells in humans and in mice. Additionally, as semi-permissive hosts, RSV-infected mice were unable to transmit virus to uninfected cage mates in stark contrast to the high transmissibility seen in human RSV infections.
- The mouse model was widely used at the priority date.
The cotton rat model
- Vaccine-associated enhancement of disease had been well-characterized in cotton rats that were subsequently challenged with replication-competent RSV as was the ability of RSV infection to induce neutralizing antibodies.
- While most of the early work in RSV was performed in the cotton rat model, this model was used less frequently from the mid-1980s onwards, with investigators preferring the mouse model.
The ovine model
- Sheep are semi-permissive for both RSV and BRSV. Sheep and lambs were considered suitable models for investigation of RSV infection and pathogenesis comparative to human infection. However, utility of the model has been limited.
The non-human primate models
- Several non-human primate models have been employed in the study of RSV infection including chimpanzees, African green monkeys, and macaques as well as the less common models of owl monkeys and baboons. Only chimpanzees were permissive to RSV infection. Diverse responses were seen among these non-human primate models with some models showing clinical disease much like human disease but making low antibody titres following natural infection or immunization or patterns of viral clearance inconsistent with that in humans. In contrast, other species would have patterns of pulmonary inflammation consistent with bronchiolitis in human infants but exhibited low viral replication or low levels of antibodies.
- There had been limited use of these models at the priority date.
Use of Non-Human Paramyxovirus Infections in the Natural Host
97. Alternative systems were also in use at the priority date as parallels to human RSV infections such as the BRSV system. Calves infected with bovine RSV evidenced viral replication, disease pathogenesis, and antiviral immune responses that closely resembled that seen in human RSV infection of young infants.
Adjuvants
- Adjuvants are substances that are added to vaccine preparations to enhance the immune response to the antigens with which they are combined: in other words, to enhance immunogenicity. When combined with the antigens, these additives can also help with delivery of the vaccine to the immune system and enhance general immune responsiveness.
- The precise mechanisms of some adjuvants remain largely undefined. However, the mechanisms of many adjuvants were understood.
- By the priority date, extensive research had been done in this field and it had been shown that many substances can achieve this immune stimulation. For example, aluminium salts and MF59 were adjuvants approved and commonly used in human vaccines. Alum, for example, acts as an antigen depot and is a mild irritant, causing the recruitment of leukocytes necessary for generation of an immune response to the site of injection.
- Ways of improving subunit vaccines by formulation with different adjuvants, and RSV ISCOMs, in which the F and G proteins are incorporated into a defined supramolecular structure of Quillaja saponin were also described in Cranage & Taylor (2005).
Assays
Immunoassays - ELISA
- An enzyme-linked immunosorbent assay (ELISA or EIA) is a solid phase immunoassay, meaning that one of the components (i.e., antigen or antibody) is fixed to a solid surface. These types of assay were used to quantify levels of a specific target (e.g., a protein) within a sample.
- In a direct ELISA, an antigen is immobilized on the surface of a well in a microtiter plate and then incubated with the antibody of interest which has been linked (conjugated) to an enzyme after which the plate is washed to remove any antibody that has not bound to the antigen. The enzyme is able to produce a detectable response by catalyzing a reaction using an enzyme-specific substrate. Common enzymes used for this purpose include horseradish peroxidase or alkaline phosphatase. More commonly an indirect ELISA is performed in order to increase the sensitivity of an ELISA. In this assay the antigen-specific antibody is not labelled, but it is detected by multiple second antibodies specific for IgG from the species of the first antibody and that are enzyme-labelled.
Neutralization assays
- A neutralization assay can be used to measure the ability of an antibody to inhibit the activation of a downstream event which occurs when a particle binds its target, such as a ligand binding to a receptor. Neutralizing antibodies are a major goal of many vaccines, and the concentration of these antibodies can be measured in a neutralization assay. In these assays, antibody-containing fluids (e.g., serum) are mixed with the vaccine antigen or a larger particle containing the antigen (such as a virus). This mixture is then applied to cells or a tissue known to be susceptible to viral infection, producing a response such as the release of a cytokine or other messenger, or which is known to be susceptible to viral infection, resulting in viral proliferation. Reduction in the defined response in the presence of increasing concentrations of the test antibody is measured. Neutralizing antibodies can also be measured in an EIA-based assay as discussed above.
- The IC50 is the concentration of antibody which is able to inhibit the defined response by 50% (Figure 8).
Figure 8: Calculation of the IC50 of an antagonist as a measure of potency.
More on protein structures
- At this point it is necessary to return to discuss further aspects of protein structures.
- In addition to the basic points discussed in paragraphs 85-97 above, the skilled structural biologist would also be aware of a further structural characterization of proteins into functional domains which may comprise portions of a single polypeptide or may form from more than one polypeptide chain. Domains can be very variable in size but are distinct functional and/or structural units in a protein, usually responsible for a particular function or interaction. The majority of proteins are multidomain proteins. For example, some proteins have a transmembrane domain, which serves to anchor a protein to a cellular membrane or other membrane-like structure like the viral envelope. In those cases, the domain of the protein that extends into the extracellular space, or the space outside the viral particle is called the ectodomain.
- Proteins can be classified according to sequence or structural similarity into different groups based on the protein families to which they belong, the domains they contain and/or the sequence features they possess.
Protein maturation and folding
- In eukaryotic cells proteins are synthesised by ribosomes in the cytoplasm (i.e. outside the nucleus). If the protein is destined to be secreted from the cell or it is to be inserted into the cell membrane, then it is targeted (by a short N-terminal signal peptide) to the endoplasmic reticulum ("ER"). The ER is a membrane bound organelle inside a eukaryotic cell that is involved in protein maturation.
- Within the ER the polypeptide(s) of the protein fold into the proper three-dimensional structure, which may involve the formation of disulfide bonds. Proteins can also be post-translationally modified. For example, amino acid residues can be glycosylated (sugar units are covalently attached to either arginine or serine/threonine residues in proteins), disulfide bonds can form between cysteine residues or polypeptides can be proteolytically processed to convert larger "pre-proteins" into smaller active proteins or to divide a long polypeptide into shorter polypeptides.
- When folded and post-translationally modified, as appropriate, mature proteins are transferred from the ER to the Golgi apparatus (another membrane bound organelle) where further modification of glycosylation occurs or polypeptides can be proteolytically processed to convert larger "pre-proteins" into smaller active proteins or to divide a long polypeptide into shorter polypeptides. Transport between the ER, the Golgi apparatus and the cell surface is achieved by a vesicular transport system. Essentially, small spheres of membrane carry both membrane associated and soluble proteins towards the cell surface membrane and fuse with it so that the membrane associated proteins end up on the cell surface and soluble proteins are secreted into the extracellular space.
- The phenomenon of protein folding is a cooperative process that involves spontaneous intramolecular interactions. This process comprises steps that stabilize a protein in a defined conformation.
- Generally, the transition of one folded conformation to another is driven by energy jumps, always directed towards the global free energy minimum conformation, which is the stable conformation.
- As proteins fold into complex quaternary structures, any changes to the amino acid sequence of a protein can have an impact on the way it folds and ultimately its biological function. [I interpolate here that that first sentence was agreed to be CGK. In response to Dr Taylor's characterisation that the CGK extended only to a rather basic level of molecular biology, Professor Weissenhorn spelt out the importance of understanding how modifications to the amino acid sequence of a protein could affect its structure in the following two sentences which were not agreed to be CGK. That lack of agreement highlighted to me that GSK/Dr Taylor's basic CGK level of molecular biology was plainly insufficient for the purposes of this case.] As Professor Weissenhorn said: it would therefore be unrealistic to manipulate proteins in this way without any consideration of how those modifications could affect the structure of the protein. That is particularly the case for the development of vaccine antigens, which rely on the presentation of particular epitopes to the immune system in order to generate a protective immune response.
- [By way of further interpolation, it will be seen that in [0018] of the Patents, they assume the Skilled Team is familiar with and able to carry out various manipulations to protein sequences. Whilst Dr Taylor's 'experienced technician' might well be able to follow those instructions and create the necessary constructs, I consider it would be unrealistic to posit the Skilled Team without the structural expertise to assess how modifications of those types or combinations of them would be likely to affect the structure of the protein.]
Recombinant nucleic acids and proteins
- When DNA from two different sources is combined together, creating sequences that would not otherwise be found in the genome, the resulting patchwork, synthetic, DNA molecule is called recombinant DNA. Moreover, DNA sequences that do not occur anywhere in nature may be created by chemical synthesis of DNA, or nucleotide changes, also known as mutations, in existing DNA sequences.
- Proteins that can result from the expression of recombinant DNA within living cells are termed recombinant proteins. Advances in immunology and protein engineering have allowed the design and production of recombinant subunit vaccines.
- By the priority date it was known that a variety of techniques were available to examine the structure of proteins. Some of these are described in further detail below.
X-ray crystallography
- X-ray crystallography is used to determine the three-dimensional structures of biomolecules such as proteins or nucleic acids at close to atomic resolution. The first stage involves growing crystals of the molecule of interest. The crystals are then exposed to an intense X-ray beam and the resulting diffraction pattern is used to produce an electron density map, which is in turn used to build a three-dimensional atomic model of an individual protein.
- For X-ray crystallography to work effectively the biomolecules need to form well-ordered crystals capable of diffracting X-rays to suitable resolutions for structure solution (generally better than 3.5 Å). Crystals comprise many repeating units, called "unit cells", which contain one or more molecules of the protein of interest. Obtaining crystals of sufficient size for X-ray analysis can be a slow and difficult process, especially because proteins are by their very nature dynamic and often expose flexible regions that hinder crystallization, that is, ordered interaction into a crystalline lattice.
- Once diffraction-quality crystals have been identified, they are exposed to a high energy X-ray beam and the resulting diffraction data is used to calculate the three-dimensional structure of molecules.
- When the primary X-ray beam passes through the crystal, some of the X-rays interact with electrons on each atom within the crystal and end up being diffracted. The particular diffraction pattern is detected due to the symmetric arrangement of atoms in a crystal and used to mathematically calculate an electron density map of the repeating unit of the crystal, which is in turn used to create the three-dimensional protein model.
- The general aim at the priority date was to obtain crystals that diffract at least to 3.5Å, which generally allowed placing of side chains into the calculated electron density map. However, interpretation of certain interactions, such as hydrogen bonds etc., need to be evaluated with caution at this resolution. High confidence resolutions start from 2.5Å and below. The calculated model is refined against the experimental electron density map and statistical analysis generally indicates the level of confidence/correctness of the atomic model. This is given as so-called "R factors", which report the error between the atomic model and the experimental data. R factors are important indicators used to evaluate the correctness of the structure for publication.
- X-rays used in traditional crystallographic experiments were typically produced by specialised laboratory X-ray generators. By the priority date, it was increasingly common to use a special type of particle accelerator called a synchrotron, which was able to produce X–rays at far higher intensities than the usual generators. By the priority date, X-ray crystallography was a well-established tool used to study the structures of viral envelope glycoproteins.
Electron microscopy
- Electron Microscopy ("EM") is a technique used to determine the tertiary or quaternary structures of biomolecules. The most common type of EM used for studying viral envelope glycoproteins is transmission electron microscopy, so-called because it involves the detection of electrons that have been transmitted through a sample. A particular preparation technique called "negative staining" is often used and was known to increase the contrast of the samples and allow visualisation of the overall shape of molecules. A drop of solution containing a heavy metal is added to the samples during fixation. Metal-stained areas appear darker on the micrograph because metal atoms diffract most of the electrons that hit them and so cannot be detected and do not contribute to the image. Areas that take up less metal staining appear lighter on the resultant image.
Homology modelling
- Homology modelling (sometimes referred to as comparative modelling) involves modelling a three-dimensional structure of a protein of interest based on its primary sequence and the known three-dimensional structure of a homologous protein which has already been determined experimentally (for example, by X-ray crystallography).
Other biophysical and biochemical techniques
- Along with X-ray crystallography and EM, a number of other techniques were used by the priority date to indirectly analyse the structure of proteins, or to measure interactions between molecules such as antigens and antibodies. These included the following.
Sucrose gradient analysis
- This technique uses ultracentrifugation to separate proteins of differing mass through a sucrose solution of increasing density. The protein sample is loaded into a sucrose solution which varies in concentration from low (near the top of the tube) to high (nearest the bottom). The sample is subjected to centrifugation, during which time the proteins within the sample migrate down the tube at different rates, dependent on their respective masses, and settle into discrete zones. This separation technique is also referred to as rate-zonal centrifugation.
Size exclusion chromatography
- Size exclusion chromatography (SEC) was a commonly established technique that was (and still is) generally used as a final step in protein purification. SEC separates molecules based on their size by filtration through a gel. The gel consists of spherical beads containing pores of a specific size distribution. Separation occurs when molecules of different sizes are included or excluded from the pores within the matrix. As a consequence, large molecules pass faster through the column than small molecules permitting the separation of proteins according to their size i.e. their molecular weight.
Liposome association assays
- Liposomes are small uni-lammelar vesicles that have been commonly used to study membrane proteins or protein interaction with membranes. Liposomes are composed of a lipid bilayer that forms a vesicle with a defined lipid composition. Protein interaction with liposomes is generally analysed by sucrose gradient centrifugation and SDS-PAGE analysis.
- The surface glycoproteins ("GPs") of enveloped viruses were understood by the skilled structural biologist at the Priority Date to play a major role in the membrane fusion process leading to host cell entry and infection. They are often trimeric molecules and composed of a receptor binding domain that mediates binding to cellular receptors and a fusion protein that catalyses the last step of virus entry via fusion of the viral and cellular membranes. The activities of receptor binding and fusion can be present in a single glycoprotein (as is the case for HIV) or two different glycoproteins acting in concert (such as certain paramyxoviruses).
- Enveloped viral GPs are generally present on the surface of the virion in a "metastable" prefusion conformation that is also referred to as the native conformation. Receptor binding induces major conformational changes, especially in the fusion protein subunit, that adopts as a consequence the stable, lowest energy state "hairpin" conformation referred to as the post-fusion conformation.
- This transition from the native to the post-fusion conformation is thought to drive membrane fusion. As at the Priority Date, the skilled structural biologist would have been aware that three different classes of enveloped virus GPs had been identified based on common structural motifs present in the post-fusion conformations of the fusion proteins. It is only necessary to mention Class I fusion proteins, which are characterized by trimers of hairpins containing a central α-helical coiled-coil structure.
- Examples of Class 1 viral fusion proteins were given by Professor Weissenhorn, including from the Orthomyxoviridae family, the Influenza A virus HA, from the Paramyxoviridae family, PIV 1-5 F, NDV F, RSV F, Human Metapneumovirus F, Measles Virus F and Sendai Virus F, from the Retroviridae family, HIV 1 gp41 and from the Coronaviridae family, the Mouse hepatitis virus S2 and the Sars corona virus E2.
Class I fusion proteins
- Class I fusion proteins are trimeric, type I transmembrane proteins synthesised as fusion-inactive single precursor proteins. The precursor proteins are subsequently cleaved into two subunits producing fusion-active proteins, and, in most cases, comprise a receptor-binding domain and a membrane-anchored fusion domain held together by covalent (i.e. inter-subunit disulfide bonds) or non-covalent interactions. Cleavage usually occurs in the Golgi apparatus via cellular proteases called furin. The furin protease has a consensus sequence specific for cleavage described as "R-X-K/R-R" or "Arg-X-Lys/Arg-Arg" where Lys and Arg are lysine and arginine residues, respectively, and X is any amino acid. Cleavage occurs immediately after the fifth or C-terminal arginine residue in the sequence.
- The N-terminal region of the fusion subunit contains a conserved hydrophobic region known as the "fusion peptide" which inserts into the target membrane during fusion. At its C-terminal end, the fusion subunit includes a transmembrane domain for anchoring in the virion surface. Another common feature of all class I viral fusion proteins is a conserved furin cleavage site.
- Well-known proteins of this class include influenza (HA), HIV-1 (env/gp-160), coronavirus (S), Ebola virus (GP) along with members of the Paramyxoviridae family, which include the PIV5 and RSV F proteins (see Table 1 above). The HA protein from influenza virus is the best characterised viral glycoprotein, and early models of the mechanism of class I protein mediated membrane fusion were developed based on structural studies of HA. Later structural studies of the fusion proteins from a variety of class I fusion proteins all pointed to a shared helical trimer-of-hairpins structure, or "core trimer", present in the post-fusion conformation of the proteins.
Influenza HA
- It was shown as early as 1975 that the influenza HA protein is expressed as a precursor polypeptide (HA0) that is cleaved by a cellular protease into two subunits: HA1, the receptor binding subunit; and HA2, the fusion protein subunit. That and other early studies indicated that cleavage was an important step in "activation" of the protein for fusion.
- Further work helped to establish the concept that conformational changes in the HA protein were required for membrane fusion, by identifying low pH as a trigger to generate the post-fusion conformation.
- In 1981, Wiley and colleagues reported the first crystal structure of HA in its native prefusion conformation. A ribbon diagram of the prefusion structure is shown in Figure 10B below, along with a schematic representation of the amino acid sequence of HA.
- In 1994, 13 years after the prefusion structure mentioned above had been solved, Wiley and colleagues published the first structure of the fusion protein subunit HA2 in the low-pH induced, post-fusion conformation, shown in Figure 10C below.
- Comparisons of these two crystal structures revealed for the first time the extensive folding or conformational changes that accompany the transition from the native (or pre-fusion) state into a fusion-active or postfusion state. The two most striking changes involve the transition of the native HA2 loop region into a helical segment (shown in red in the figure below) exposing the otherwise sequestered fusion peptide sequence (shown in yellow in the figure below) and the reversal of the chain towards the end of the central triple-stranded coiled-coil structure.
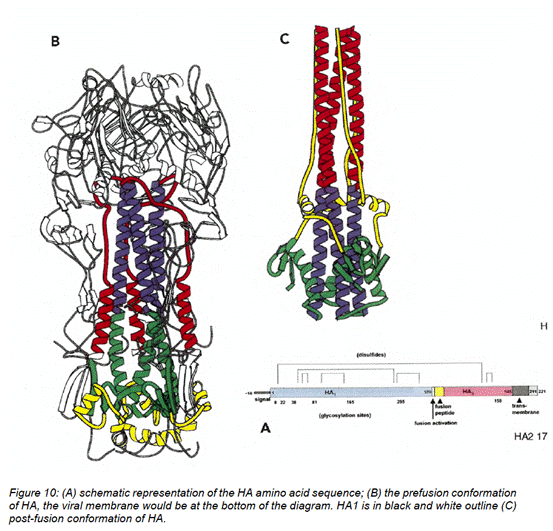
- Lest it be thought that these crystal structures were too niche to be CGK, it should be noted that this figure is set out in one of the introductory chapters in Fields: Chapter 3: Principles of Virus Structure: Fig 28 in the colour plates and Fig. 3.38 on page 85.
- It was not at that stage clear how the conformational transitions could bring membranes close enough to induce membrane fusion, because the structure of the prefusion conformation lacked the C-terminal region connected to the transmembrane region. Later structural work confirmed that the C-terminal folds back onto the N-terminal coiled coil, placing both the transmembrane anchor and the fusion peptide at the end of a rod-like structure - this was first established for the HIV-1 envelope glycoprotein subunit gp41.
- Further work showed that the influenza HA2 subunit spontaneously folds into the postfusion conformation in the absence of the receptor binding domain HA1, and that the complex of the HA1 and HA2 subunits is metastable. Therefore, the induction of the fusion reaction leads to the most stable conformation of HA2, a process which involves extensive refolding.
Common structural features with other class I fusion proteins
- Since the late 1980s, structural similarities between viral membrane fusion proteins had been suggested based on the presence of predicted coiled-coil regions adjacent to the fusion peptide region, within the fusion subunit.
- Throughout the 1990s, crystal structures from a variety of distantly related viral fusion proteins highlighted structural similarities to the post-fusion influenza HA2 structure mentioned above. They all contained central triple-stranded coiled-coil regions with the C-terminal sequences folded back in an anti-parallel fashion with respect to the coiled coil, so that the N-terminal region (i.e. the fusion protein) and the C-terminal region (i.e. transmembrane anchor) are positioned next to each other. As explained above, that rearrangement of the N- and C-terminal regions was thought to have major implications for membrane fusion.
- Activation of the glycoprotein allows the fusion protein to be anchored in both the viral membrane (via the transmembrane region) and the host cell membrane (via the fusion peptide) simultaneously, bridging the viral and cellular membranes. The two helical parts will then refold like a jack knife and bring the membranes into close apposition leading to the post-fusion conformation that has the fusion peptide and the transmembrane region anchored in the same membrane. Several examples of post-fusion structures of class I fusion proteins known by the Priority Date are shown at Figure 11 below.
- Although no complete crystal structure of the prefusion conformation of a class I fusion protein ectodomain, other than influenza HA, was obtained until 2006 (see Yin below) the structural similarity of all the known fusion core structures (as illustrated in Figure 11) led the field to assume that they were all the product of conformational changes from the pre to the postfusion form, in analogy to influenza hemagglutinin.
- Following early work on influenza HA, studies on another class I viral fusion protein, the HIV-1 envelope protein, provided further information about the structural and functional role of these proteins in viral membrane fusion.
HIV Envelope
- The HIV envelope ("Env") is synthesised as a precursor protein called gp160, which is subsequently proteolytically cleaved into the receptor binding subunit gp120 (analogous to influenza HA1) and the fusion subunit gp41 (analogous to HA2). Those two polypeptides associate together into the native envelope glycoprotein, composed of three gp120-gp41 dimers. Unlike influenza HA, though, the gp120 and gp41 do not associate covalently and there are no disulfide bonds holding them together. As a result, the Env protein is a lot less stable and leads to a process known as viral "shedding" involving dissociation of gp120 from the Env trimer.
- The HIV-1 envelope glycoprotein has been studied extensively since the discovery of the virus in the early eighties. Along with colleagues in the Wiley lab, Professor Weissenhorn's group published the first high-resolution structure of the core trimer of the gp41 subunit was published in 1997 (as a result of a collaboration between the Wiley lab and Professor Weissenhorn's lab), together with another paper by Peter Kim and colleagues. The gp41 construct used in that study was fused with a GCN4 domain at its N-terminus, in place of the fusion peptide, in order to solubilize the protein for crystallization. The work on gp41 was then followed by structure determination of the receptor binding domain called gp120 in 1998, and many subsequent structures in complex with newly identified neutralizing antibodies, eventually establishing the field of reverse vaccinology.
- The importance of the placement of the fusion peptide in close proximity to the membrane anchor was first recognized in the HIV-1 gp41 crystal structure. That discovery allowed Professor Weissenhorn's group to propose a potential mechanism for a generalized fusion model, based also in part on the structural homology between the gp41 and the HA2 post-fusion structures. Those structures validated a model for membrane fusion whereby the conformational changes induced by receptor binding lead to the refolding of the fusion protein, a process that places the fusion peptide next to the transmembrane region in the final, lowest energy state conformation upon completion of membrane fusion.
- Despite some modifications, the model proposed was by the Priority Date generally accepted for all class I membrane fusion proteins. This point is confirmed by the distinct similarity between the figure (shown below) which Professor Weissenhorn reproduced in his report taken from the paper published in 2000 by Skehel & Wiley (Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531-69) and the plate from Cane I set out under paragraph 149 above. A very similar model is also set out in Fields Chapter 3, colour plate IV and Figure 3.30 on page 86: 'Fusion by class 1 viral fusion proteins'.
- In the first step, proteolytic cleavage of the glycoprotein precursor molecule transforms the glycoprotein into a 'metastable' conformation. Activation of the fusion protein by its trigger, which is commonly either receptor binding or changes in pH, induces a conformational change in the fusion protein subunit. As a consequence of activation, the N-terminal fusion peptide is extended towards and anchored in the host cell membrane.
- Evidence suggested that this conformational change leads to a transient intermediate, termed the pre-hairpin structure, which is a major target for fusion inhibition. This stage may be somewhat reversible in the absence of membranes, however upon insertion of the fusion peptide into its target membrane, the fusion protein is committed to the fusion reaction. This is followed by further refolding of the fusion protein, resulting in the helical hairpin structures, a process generally thought by the Priority Date to pull the two membranes into close proximity leading to membrane fusion.
- The process of lipid bilayer membrane fusion is common to all enveloped viral entry pathways. As cell membranes composed of lipid bilayers repel each other and do not fuse spontaneously, a substantial energy barrier must be overcome for fusion to take place. Within that context, viral fusion proteins control the fusion process by generating initial contact between the two membranes via extensive conformational rearrangement of the viral fusion proteins, which will bring the membranes into close apposition thereby lowering the energy barrier required to join the two membranes together.
- By the Priority Date, the membrane fusion reaction was known to be explained by the generation of lipid intermediate structures controlled by the conformational rearrangements of the viral fusion proteins. The refolding of the fusion protein brings the membranes into close apposition, which allows mixing of the lipids of the outer lipid bilayers leading to a structure named "stalk" that develops into a "hemifusion diaphragm", which breaks to open the fusion pore. The latter expands and eventually fuses the complete viral membrane with the cellular membrane.
Paramyxoviruses
- Like other Class 1 fusion proteins, paramyxovirus fusion proteins are synthesised as an F0 precursor and cleaved by a furin protease enzyme before activation. Cleavage results in F1 and F2 subunits, which are covalently linked by inter-subunit disulfide bonds. A hydrophobic fusion peptide is positioned at the N-terminal end of the F1 subunit with a transmembrane anchor at the C-terminal end, in structural analogy to the fusion subunits of influenza (HA2) and HIV (gp41). The F1 subunits also contain 7-residue (i.e. heptad) repeat regions designated HRA and HRB.
- Comparisons of the amino acid sequences of paramyxovirus fusion proteins had revealed a relatively low level of sequence similarity, except for the fusion peptide, which has a conserved sequence of up to 90% identity. Despite those sequence differences, comparing the placement of certain residues within the sequences indicated a similar structure for all F proteins of that family. The structural biologist would have known that RSV differed from other paramyxoviruses in that it comprised two furin cleavage sites instead of one, and that cleavage resulted in the removal of an intervening 27-amino acid region called pep27. Professor Weissenhorn added that even if they were not aware of the nuances of the RSV F protein structure, the Skilled Structural Biologist would know to look it up in the relevant chapter of Fields Virology.
- The structural biologist would also have known that efforts to characterize the structure of the paramyxovirus fusion protein F were underway. The first crystal structure of the core trimer of the Parainfluenza Virus 5 fusion protein (PIV5, formerly known as Simian virus 5, or SV5) that was resistant to further proteolysis (and so likely in the stable, postfusion conformation) was published in 1999. Strong similarity with the previously characterized HA2 and gp41 crystal structures confirmed that the PIV5 structure represented the post-fusion conformation of that protein. A crystal structure of the RSV core trimer also showed striking similarity to the post-fusion conformations of HIV and influenza HA.
- As part of his explanation of the development of these structures, Professor Weissenhorn referred to an issue which arose from the subsequent crystal structure of the proteolytically cleaved fusion protein (residues 33–105 and 171–454) from NDV which was first interpreted as representing the prefusion conformation of the NDV F protein. However, the crystal structure was produced from purified F0 glycoprotein which the authors noted had been proteolytically cleaved prior to crystallization, and that likely had an effect on the native, metastable F0 structure. It could be speculated that the proteolytic cleavage facilitated the spontaneous transition of the prefusion F to post fusion F conformation. Indeed, a combined model of the six helical bundle PIV5 F core structure with the NDV F0 structure suggested that both are part of the larger complete post fusion F structure. I was not convinced that the Skilled Structural Biologist would have known this level of detail as part of his or her CGK, but that does not matter because of what follows.
- The issue was resolved in 2006 following the publication of a landmark paper by Ted Jardetzky and colleagues, which disclosed the prefusion, native structure of the fusion protein from PIV5 (this is the Yin prior art paper). In order to stabilize the prefusion conformation of the PIV5 F for crystallographic characterization, the authors fused the F protein with a C-terminal trimerization domain called GCN4 in place of the transmembrane region. This paper was particularly important in the field, because it not only reported the first crystal structure of the prefusion conformation of a paramyxovirus F protein (and only the second class I fusion protein prefusion structure, following influenza HA in 1981), it also demonstrated that a trimerization domain could be used to stabilize the F protein in the prefusion conformation by countering the instability caused by removal of the native transmembrane domain. The skilled structural biologist working on or interested in viral envelope glycoproteins would have been aware of this paper and, to the extent they were not aware of specific details but wanted to find out more, would have known where to look it up in order to do so.
- A structure of the post-fusion conformation of a closely related paramyxovirus F protein, PIV3, had already been published by the same lab a year earlier and was the subject of considerable interest in the field, which the structural biologist would have known ("Yin 2005"). Both the PIV3 and NDV crystal structures were based on "uncleaved" versions of fusion proteins, whereby furin cleavage into two separate subunits was prevented. In addition, the transmembrane domains had been removed so as to allow secretion of soluble, "anchorless" proteins for crystallization. As described above, by that time it was well established from work in influenza and HIV that cleavage of class I fusion proteins was a trigger for "activation" of the protein and their conformational switch to the stable, post-fusion state. It was therefore surprising to the field that these uncleaved, soluble proteins were in the postfusion form. One explanation for that result was provided by the authors of Yin (and set out in Fields Virology at Chapter 41, page 1469, in the passage bridging the columns) and would have been understood by the structural biologist to be one of the key messages from that paper - that the transmembrane domain has a role in stabilizing the prefusion conformation of the F protein and so its removal in the soluble form of the protein means it spontaneously folds to the stable, post-fusion conformation.
- Comparing the pre- and post-fusion crystal structures allowed a greater understanding of the molecular details of the conformational transitions from the native state to the post-fusion state (which until then had only been determined for influenza HA protein). Combined with other molecular and biochemical work on the paramyxovirus F proteins, the authors were able to propose an updated model for F protein mediated membrane fusion.
- In the first step, the HRB helices separate apart to form an 'open-stalk' intermediate form. The HRA regions then form into coiled-coil helices and the fusion peptide translates towards the target cell membrane in what is known as a "pre-hairpin intermediate". The HRB regions then refold in a movement that most likely moves the two membranes in close apposition, followed by the assembly of the final six helix bundle which completes the conformational change and merger of the two membranes. See Figure 14 below (which is Figure 41.19 in Fields, Chapter 41 and taken from Yin):

- Professor Weissenhorn said, correctly in my view, that the publication of the prefusion structure of PIV5 was therefore a particularly important advancement in the field and would have been read with interest by the skilled structural biologist working on the structural biology of viral proteins. Despite only being published in 2006, the Yin paper had such an impact in the field that it was referred to extensively in the latest edition of the CGK textbook Fields Virology published a year later (see for example page 87 in Fields Chapter 3 and pages 1466 to 1471 of Fields Chapter 41, in which Yin is ref 422 and Yin 2005 is ref 421).
- As explained above, by the Priority Date the skilled structural biologist knew that enveloped virus fusion glycoproteins are predominately in a metastable prefusion conformation on the virion surface, which folds to the stable post-fusion conformation. The skilled structural biologist would also have been aware that in order to produce soluble glycoproteins for structural studies or immunization experiments in a vaccine context, a general strategy employed at the time was to express only the ectodomain of the fusion glycoprotein (as was the case for the work on PIV3 and NDV, described above) i.e. lacking the transmembrane and cytoplasmic regions of the protein. One of the points that the skilled biologist would have understood from the Yin work is that the absence of the transmembrane domain, which contributes to the stability of the prefusion trimer, renders the glycoprotein very often more labile or more prone to switch spontaneously to the post-fusion conformation. That also explained why obtaining sufficient amounts of the fusion protein in the prefusion conformation for structural characterization proved very difficult - the influenza HA protein being the only exception, as it remained stable in the prefusion conformation after enzymatically cleaving the ectodomain from the virus surface, and could be crystallized without any additional modifications.
- In order to stabilize the trimeric and/or native prefusion conformation and prevent its switching to the post-fusion state, the structural biologist would have understood that different strategies could be employed, a number of which had already been developed and tested in HIV-1 with the aim of preserving the native Env trimer conformation. The most common one was the addition or fusion of a small trimerization domain in place of the transmembrane domain. The structural biologist would have been aware of a number of common and well-characterized trimerization domains, such as trimeric versions of GCN4, a coiled-coil domain described by the Kim lab in the early 1990s, or the foldon domain.
GCN4 domains
- The GCN4 (sometimes referred to GCNt or GCN4t) domain is a short 30 residue long peptide that is derived from the yeast transcription factor GCN4. It can adopt dimeric, trimeric and tetrameric oligomeric forms depending on its sequence. It is a typical coiled-coil domain composed of seven amino acid repetitions (designated abcdefg), called heptad repeats, in which the a and d positions of the heptad motif are generally occupied by hydrophobic residues positioned at the interface between helices. Hydrophobic interactions between the a and d residues form the core of the oligomeric interaction. The residues at positions e and g are solvent-exposed and tend to be polar residues which allow electrostatic interactions between helices. A wheel representation of three helices in a coiled coil zipper is shown at Figure 15 below:
Figure 15: Helical wheel diagram of a trimeric leucine zipper coiled-coil. Inter-helical hydrophobic (a/d) and electrostatic (e/g) interactions are indicated by dashed lines.
- The trimeric form of the GCN4 domain was developed by the Kim lab in 1994, in which the leucine residues at positions a and d of the heptad repeat were changed to isoleucine. The trimeric structure of the isoleucine zipper is shown below.

- The helical nature of the GCN4 domain means that it can be fused in-frame with the sequence of the protein of interest. Fusing "in frame" means joining the native and GCN4 domain DNA sequences in such a way that the heptad repeats of the resulting fused sequence remain intact.
Foldon domains
- The skilled structural biologist would have known of the foldon domain, a different type of domain composed of 27 amino acids that fold into a small beta-hairpin configuration that trimerizes spontaneously. It is a naturally trimeric, highly soluble protein derived from the bacteriophage T4 fibritin protein. It folds as an independent domain with a hydrophobic interface, which allows its fusion to any protein partner to stabilize its trimeric oligomeric state. Unlike the GCN4 domain, which contains coiled-coil regions often found in nature, the foldon sequence is not common in genomes. By the Priority Date, the skilled structural biologist would have been aware that foldon domains had been added to recombinant viral envelope glycoproteins, such as the HIV-1 env in order to stabilize them in trimeric form, and would consider them part of the standard tool kit for stabilizing such glycoproteins.
- The trimerization domains are either directly fused to the C-terminal ends or fused via linker sequences. Both domains would be well-known to the skilled structural biologist working on viral envelope glycoproteins. For example, the Sodroski lab fused a GCN4 domain to the C-terminal end of an uncleaved polypeptide containing gp120 and the ectodomain of gp41, commonly known as gp140, in order to generate more stable, native-like trimers, and showed that soluble, uncleaved gp140 molecules containing a foldon domain were more stable to heat and reducing conditions than the GCN4 construct. The authors suggested that these molecules assume conformations distinct from that of the post-fusion, six-helix bundle. A foldon domain was also used to stabilize the native rabies virus envelope glycoprotein.
Other modifications
- Other stabilization techniques that would be known to the skilled structural biologist to have been employed in HIV-1 included disruption of the proteolytic cleavage site between gp120 and gp41 (equivalent to the lack of furin cleavage sites in the PIV5 and NDV fusion proteins, described above), and the introduction of cysteine residues that form gp120-gp41 inter-subunit disulfide bonds. The structural biologist would have been aware of those approaches and understood them to have had general application to other viral envelope glycoproteins.
- Early attempts at engineering a disulfide bond between gp120 and gp41 to prevent gp120 shedding from cleaved Env trimers, was not sufficient to prevent trimer disassembly. The additional introduction of a proline at the Ile559 position (I559P) then led to the first stabilized prefusion Env trimer, named gp140 SOSIP. By the Priority Date, the conformational stability had been confirmed by a range of biochemical, biophysical and immunological assays. Together, this work showed the importance of structural analyses in the characterization of the conformational states of enveloped virus glycoproteins.
- Another widely used technique that would have been well known to the structural biologist to increase the stability of recombinant fusion proteins was mutation or removal of the cleavage sites. As discussed above, early studies on influenza HA had established the principle, common amongst class I fusion proteins, that cleavage by furin-like protease was an important activation step before the conformational change that leads to membrane fusion. Removal of the protease cleavage sites, either by mutation of the furin recognition sequence or deletion from the DNA sequence, to produce so-called "uncleaved" proteins was a commonly used approach in the field to potentially increase or maintain the stability of the protein for crystallography or other structural studies dependent on conformation.
- The structural work described above not only helped to explain the mechanism of surface glycoprotein-mediated membrane fusion, but also pointed to the immunological importance of the different conformations of the fusion proteins. By the Priority Date it was well recognised that generating recombinant, stable forms of viral envelope glycoproteins could be important both for structural studies - and particularly for viruses such as HIV-1 where efforts to crystallize the prefusion structure of the Env protein had so far been unsuccessful - and for use as vaccine antigens.
- As an example, Professor Weissenhorn cited the crystal structure of influenza virus glycoprotein HA which indicated for the first time the importance of the native, prefusion conformation for the identification of major antigenic sites that are associated with different influenza virus strains.
- Furthermore, the structural biologist would have been aware of work on HIV-1 which established that most of the antibody responses directed towards the HIV Env are non-neutralizing and that the few monoclonal antibodies that potently neutralize HIV-1 all recognize epitopes exposed on the native Env complex. The structural biologist would have known that the post-fusion form of gp41 was also immunodominant (i.e. the immune response is skewed towards antigenic sites on the post-fusion form of the protein), and although antibodies to the post-fusion form were common in the sera of HIV-1 infected humans, post-fusion specific antibodies are largely non-neutralizing and did not bind to the native HIV envelope complex.
- The structural biologist would have appreciated that this finding was not particularly surprising. The concept of enveloped virus glycoproteins existing in at least two conformations, the native prefusion conformation and the post-fusion conformation, had by the Priority Date been well established and indicated indirectly that antibodies targeting the post-fusion conformation are more likely to be non-neutralizing because they recognize a conformational state of the glycoprotein that is formed only after entry - hence why no protection can be provided.
- Conversely, the structural biologist would also have appreciated that antibodies, which target the prefusion conformation would likely be more effective at preventing membrane fusion and therefore viral infection and disease progression. Interaction with the prefusion conformation would be more likely to disrupt the activity of the glycoprotein or interfere with it adopting the postfusion conformation. As a consequence, any inhibitory action of antibodies would likely prevent virus fusion with the host cell membrane.
- They would also have been aware of unsuccessful attempts to generate neutralizing antibodies using recombinant monomeric forms of the HIV-1 envelope glycoprotein. That finding, together with the fact that neutralizing HIV-1 antibodies appeared to recognise epitopes on the native forms of the HIV Env protein, also supported the idea that protein antigens which more closely represented the native, virion-bound trimeric structure of the envelope glycoprotein, and which were important for driving the fusion process, could serve as better immunogens. This is because the monomeric forms did not represent the native trimeric structures present on the surface of the virion, and so would not present many epitopes specific to the neutralizing antibodies.
- It will be appreciated that in my recitation of the Technical Background above, I essentially resolved the issue I identified at [82i)] above, and I have touched upon aspects of the other disputes identified in [82], but before I reach conclusions on those other critical issues it is necessary to summarise how certain issues developed, particularly by reference to the written evidence of Drs Johnson and Taylor, and then some criticisms of the way the evidence emerged.
- In order to understand parts of what follow, I will add this diagram taken from the Calder paper. Although this paper was well-known, it is unlikely that every aspect of it was CGK. However, what certainly was CGK was Calder's characterisation of the cone and lollipop-shaped rods and the positions of the antigenic sites, as shown and explained in Figure 4 from the paper, together with its rubric. Even if the Skilled Team did not have this diagram in mind, they would know where to find it:
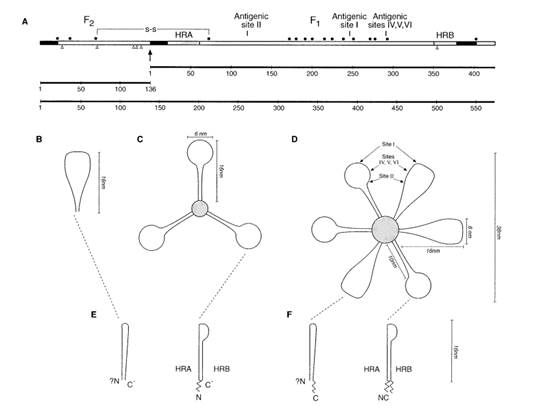

- Although I have already set out my conclusions as to the makeup of the Skilled Team and the relevant Technical Background, what I have set out in this section provided context for those decisions. It also provides context for my consideration of the primary evidence of obviousness and especially for GSK's case on secondary evidence.
- The Grounds of Invalidity contain Pfizer's allegations of obviousness over various pieces of prior art, set out in a conventional manner. GSK's Defence met the allegations of obviousness with a bare denial. Neither at that initial stage, nor subsequently did GSK plead that they relied on a case of secondary evidence to rebut the allegations of obviousness.
- As already indicated and as developed in the first reports of Dr Johnson and Professor Weissenhorn, it was Pfizer's case that the Skilled Team comprised a Skilled Vaccinologist and a Skilled Structural Biologist. By contrast, Dr Taylor considered that specialist structural biology expertise was not required, and that the Skilled Vaccinologist would have a sufficient knowledge of structural matters. This was (at least in part) based on her understanding that a structural biologist is a person interested in the elucidation of protein structure by methods such as X-ray diffraction. For those two reasons, Dr Taylor had in mind a very much narrower role for structural biology than Dr Johnson and Pfizer.
- This was not the only cause of the narrow approach taken by Dr Taylor. Her Skilled Team and her evidence was very much focussed on RSV, almost to the exclusion of other paramyxoviruses. In her reply report, Dr Taylor said '..the RSV vaccinologist would not have followed multiple paramyxoviruses closely.' Since both Yin and the Jardetzky art discuss other paramyxoviruses, her primary evidence on obviousness was dismissive. For example, she discussed the case based on Yin in the following way in her first report:
'Yin is not specifically directed to RSV nor is it concerned with vaccine development. It would not provide an obvious RSV vaccination strategy to the Skilled Team. It is a report of F protein structures of PIV5 from the perspective of its structural biology. It discusses the conformation of pre and postfusion F proteins of PIV5 and PIV3, respectively. The Skilled Team would know such conformation changes and their mechanisms were not well understood at the Relevant Dates and that Yin is trying to develop that understanding. Yin discloses nothing which would be read as being relevant to vaccination against RSV and gives the Skilled Team interested in developing vaccines no reason to produce a RSV F protein stabilised in the pre-fusion conformation.
Furthermore Yin does not expressly teach how to stabilise the RSV F protein in the prefusion conformation and I do not believe the Skilled Team would assume that, because PIV5 had been stabilised, a similar approach would necessarily work with the RSV F protein. That would require further research.
For these reasons I do not consider that the Yin paper makes the contents of the Relevant Claims obvious.'
- In consequence of the issues over the membership and skillset of the Skilled Team, Dr Taylor's approach to the CGK was narrow as a result. On Pfizer's case therefore, the Statement of Agreed CGK was deficient in a number of important respects.
- This was not the only driver however for Dr Taylor's approach to the CGK. She took particular issue with certain key paragraphs in the first report of Dr Johnson, [124] and [125] in particular which I have to address in greater detail later. Dr Taylor made her position very clear:
'The common general knowledge did not include the approach of developing as a vaccine an F protein stabilised in the prefusion conformation.
The idea of vaccinating with the F protein in its prefusion conformation had not occurred to me at the Relevant Dates. I was aware that the F protein had two conformations and I was familiar with the Calder paper. I had by the Relevant Dates been focused on the problem of RSV vaccination for over 25 years but at no point did I make a connection between the two conformations and the design of HRSV (or BRSV) vaccines. Furthermore I have no recollection of having discussed this idea with others working in the field or having heard this being discussed.
I have addressed my mind to whether my thinking was typical of people working in the field at the Relevant Dates and believe it was. There are numerous review articles in the field around the Relevant Dates and, as far as I am aware, none of them draw attention to the potential relevance of different forms of the F protein in developing a vaccination strategy and none suggest the approach of vaccinating with a subunit vaccine comprising the F protein stabilised in the prefusion conformation.'
- In these paragraphs, Dr Taylor seems to me to be addressing herself to a case of obviousness over CGK, which Pfizer did not advance. By contrast, in her first report, Dr Johnson had explained her views that the Patents were obvious over each piece of prior art. However, as GSK pointed out, all her views on obviousness were clearly founded on her views as to CGK and particularly the points set out in [124] and [125] of her first report.
- Dr Taylor's reply report (Taylor III) is relatively short - 17 pages of text. Her response to Dr Johnson's first report identifies the key issues:
i) The Skilled Team would not include a specialist in structural biology;
ii) The RSV Vaccinologist would not following multiple paramyxoviruses closely;
iii) As already indicated, she strongly disagreed with Dr Johnson's [124] and [125] on the CGK and stated in terms that:
'The relevance of prefusion and postfusion conformations of the RSV F protein to a vaccination strategy was not part of the Skilled Team's thinking at the Relevant Dates, as reflected in the review articles which I referred to at Taylor 1 paragraph 5.55.'
- Having cited and exhibited a range of those review articles, Dr Taylor concluded:
'None of the review articles above nor any of the other review articles before the Relevant Dates that I reviewed in preparing Taylor 1 listed in Exhibit GT-11 refer to the prefusion or postfusion RSV F protein as being something to consider in vaccine development.'
- She then went on to say (in paragraph 2.22) that the earliest review article she had been able to identify that 'refers to the prefusion or postfusion RSV F protein in the context of RSV vaccine development is by Dr Julia Hurwitz, published in October 2011. Part of the abstract conveniently summarises the article:
'Despite half a century of dedicated research, there remains no licensed vaccine product. Herein are described past and current efforts to harness innate and adaptive immune potentials to combat RSV. A plethora of candidate vaccine products and strategies are reviewed.'
- As Dr Taylor said, the various existing (i.e. pre-priority) subunit vaccine projects are discussed. Dr Taylor's observations on Hurwitz continued as follows: 'In her summary on purified F protein she explains that despite modest neutralizing antibody activity: 'Purified F proteins remain a topic of interest, with recent attention paid to the protein's postfusion structure [169]'. Reference [169] is to Swanson 2011, which Dr Taylor noted had been mentioned by Dr Johnson in her first report. This mention was in the context of Pfizer's insufficiency plea concerning whether the Skilled Team would be able to determine whether an RSV F protein is stabilized in the prefusion conformation. In that context, Dr Taylor referred to Swanson 2011 and McLellan 2013.
- As regards her evidence in reply on Yin, Dr Taylor denied that Yin had any particular significance for a vaccination strategy to the unimaginative Skilled Team, for the reasons she had already given. She emphasised her disagreement with Dr Johnson's focus on the antigenic differences between the pre- and post-fusion forms of the F protein. For example:
'I also disagree further in paragraph 201 that "it would have been clear to the RSV Vaccinologist that immunizing with an RSV F protein that adopts a postfusion conformation might not lead to induction of antibodies that recognize the prefusion conformation". As I have already stated RSV vaccinologists at the priority date were not addressing themselves to the question of antigenic differences between the pre- and post-fusion forms of the F protein. Yin does not address this subject. It is only with hindsight that we know that there are relevant antigenic differences between the two forms of the F protein. Furthermore I am not aware of any theory in the field at the Relevant Dates that binding of neutralising antibodies to the prefusion conformation of RSV F "would prevent transition to the postfusion state and block fusion of the membranes, thereby blocking virus infection". I do not recall any such theory being proposed or discussed.
...
'The difference between Dr Johnson and me is that she contends that a prefusion form of the virus was an obvious target and would have been seen to be advantageous. I disagree for the reasons I have given.'
- Thus, in her reply report, Dr Taylor did not concede any ground. Neither did Dr Johnson. On the written evidence therefore, it was clear that the major battlegrounds for trial concerned the membership and skillset of the Skilled Team and their CGK.
- The reference to Hurwitz in Dr Taylor's reply report was the foundation for GSK's case on secondary evidence. It was explained as follows in GSK's Opening Skeleton:
'112. In support of its case that the claims are not obvious in the light of Yin GSK rely upon the secondary evidence of what was being reported in the literature and Dr Taylor's evidence. The contemporaneous literature around the priority date does not address the option of vaccinating with the prefusion form of the F protein. If Dr Johnson is right and at the priority date, even before reading Yin it was known that there was a prefusion and postfusion form of the F protein and it was known or obvious that it was advantageous to vaccinate with the prefusion form, one would expect that to be stated somewhere in the literature. The first time this is mentioned (other than in the Patents) is in 2011 (see Taylor III paragraph 2.22).'
- Since Hurwitz 2011 was only mentioned in Dr Taylor's reply report, the secondary evidence issue was not really explored in the written evidence. However, that issue seemed to me to account for extensive cross-examination of Dr Johnson, who gave her evidence first. I did not find this a satisfactory way to proceed, because it meant that the evidence which Dr Johnson wished to give on the issue could only emerge in cross-examination. Furthermore, a tightly controlled cross-examination where the witness is taken through a considerable number of passages from various papers at speed (which is what occurred) can inhibit that process. Accordingly, when I come to consider GSK's secondary evidence case, it is necessary to pay close attention to certain answers which were given by Dr Johnson.
Her instruction
- GSK and Pfizer took a fundamentally different approach to the task of instructing their experts. This was a consequence of the case each side was running. GSK's technical expert witnesses were prevented from communicating with each other and took no steps to take into account the knowledge of other members of the team.
- As I have already mentioned, Dr Taylor's written evidence suffered from a very narrow approach. I doubt this was (wholly or partly) her fault, it being far more likely that she was encouraged to take such a narrow approach by GSK's lawyers. There were two main aspects to this. First, Pfizer were critical of the 'siloed' approach taken by GSK - Dr Taylor did not and was not asked to take any account of the approach and CGK of the structural biologist. Pfizer's second point was more subtle: they contended that her narrow approach also resulted from Dr Taylor using her own experience and that of her team at Pirbright, which focussed on immunology and was more animal based than that of the Skilled Team, as a proxy for the Skilled Team. There was force in both points.
GSK's criticisms of the way Dr Taylor was cross-examined.
- It is convenient to deal with these points here. I take the four criticisms in turn.
- GSK's first criticism was that during much of the cross-examination of Dr Taylor, it was unclear whether the cross-examiner was dealing with the CGK or what was obvious over the CGK. He was accused of moving from document to document, plucking out sentences and linking them together. I was warned that careful reading of some passages was required.
- In preparing this judgment I have had to review with care the whole of the transcript of all the expert evidence, but with particular attention to the oral evidence given by Drs Johnson and Taylor. This was necessitated by two important parts of GSK's case, the first being their challenge on the interrelated points as to the identity of the Skilled Team, whether it included a structural biologist, and CGK; and the second being the case which GSK developed as to secondary evidence of non-obviousness. That first aspect of GSK's case required Pfizer to spend a lot of time in cross-examination going through what were acknowledged to be the CGK textbooks to establish what was CGK. Since GSK's real case on secondary evidence was not pleaded and hardly developed in Dr Taylor's evidence, that second aspect of GSK's case meant that GSK's Counsel had to try to build the case through a lengthy period of cross-examination of Dr Johnson, by reference to a large number of published papers. Although I discuss the topic of secondary evidence in greater detail below, the reason for mentioning these points here is to note that the accusation levelled against Mr Moody-Stuart's cross-examination (of moving from document to document, plucking out sentences and linking them together) was, from my perspective, largely if not equally true of Dr Turner's cross-examination.
- I consider it was clear when Dr Taylor was being cross-examined about the CGK and when she was being cross-examined about the obviousness allegations. It should be remembered that Pfizer did not advance a case that the Patents were obvious over the CGK, despite the fact that GSK appeared to characterise that as being the main allegation in Pfizer's case.
- GSK's second criticism comprised four more granular points relating to the cross-examination of Dr Taylor, where GSK allege that questions were put to Dr Taylor on a false basis. I address the four points in turn.
- The first concerned the characterisation of a paper by Dormitzer et al. The criticism was that Mr Moody-Stuart incorrectly but inadvertently informed the witness that a passage in the paper concerned 'paramyxovirus F' whereas in fact it related to HIV. I accept this was not done deliberately. The point was corrected in re-examination, but I accept that may not remedy the effect of a mis-characterisation in the flow of cross-examination. Having reviewed this passage of cross-examination, I do not consider this point to be material. Furthermore, one of the points being put was that, as pointed out in the CGK textbooks, it was appropriate to draw analogies with other Class 1 fusion proteins, which included HIV.
- The second point concerned an allegation that Pfizer's Counsel had put to Dr Taylor that certain matters were not dealt with in her reports, when GSK's Counsel submitted they had been. I have reviewed the relevant passage of the transcript (T4/800/20-803/17, which was a 'wrap-up' after Counsel had taken the witness through the textbook chapters) and also the re-examination on this point. It is true that in her first report, under the heading 'Sources of CGK', Dr Taylor said she had refreshed her memory of what was happening in the field using the two well-known textbooks. She referred to and exhibited Chapters 1, 2 and 8 of Cane and Chapters 15, 41, 42 and 46 of Fields. It is clear from other passages in her reports that she certainly did not accept the contents of all those chapters were CGK. Indeed, as I have already mentioned she took the view that the Skilled Team did not follow other paramyxoviruses closely, she rejected the analogy drawn by Dr Johnson between PIV and RSV and she adhered to her view that no specialist structural expertise was required in the Skilled Team.
- At the conclusion of this passage of cross-examination are three answers which Dr Taylor sought to correct at the start of the second day of her cross-examination. I address those corrections in the next section. However, I regard the passage of cross-examination as entirely legitimate. Counsel extracted concessions that the virology of RSV was viewed as relevant to PIV at the priority date and vice versa. These concessions were inevitable in the light of the CGK textbook chapters. One consequence is that I do not regard this passage of cross-examination as indicating how tired Dr Taylor was or that she was unable to concentrate on the questions, as Counsel for GSK suggested.
- To the extent that it might be said that Dr Taylor had 'dealt with' these issues in her written evidence, it had been done in such an equivocal way that Counsel for Pfizer had to put these points to her, and, in my view, it was done fairly.
- GSK's third point concerned some cross-examination on a paper by Boon et al. This paper is headed 'Meeting Report - Viral Vaccine meeting held in Barcelona, October 25-28, 2003'. As stated in the introductory paragraphs:
'The meeting brought together leading experts from all over the world, working on the development and use of vaccines against the most important virus infections of humans and animals.'
- The scientific advisory committee included some names well-known in the field, such as James Crowe, José Melero and Peter Openshaw. The paper follows the structure of the meeting, divided into 10 sessions, 'each presenting the state of the art knowledge and ongoing development in key areas of virus vaccine related research'. Part 2 of the paper concerned RSV and included a list of potential vaccine candidates for RSV as including adenovirus recombinants, immune stimulating complex (ISCOM) preparations and sub-unit vaccines, as well as LA virus vaccines. Part 7 concerned Vectored and DNA-based viral vaccines. The cross-examination focussed on the final paragraph of that section which reads as follows:
'José Melero, using state of the art technology, has identified the fusion properties and fusion sites of RSV fusion protein (F-protein) [31]. Future research will be aimed at the development of compounds or antibodies capable of binding to the fusogenic intermediate of the F-protein.'
- Footnote 31 was to a 2002 paper by Ruiz-Arguello et al entitled 'Effect of proteolytic processing at two distinct sites on shape and aggregation of an anchorless fusion protein of human respiratory syncytial virus and fate of the intervening segment.' The authors were a combination of the Melero and Calder groups.
- GSK's complaint was that this passage was put on the basis that Professor Melero had said at a conference in 2003 that antibodies which interfere with the fusion of F would be a desirable target and cited this question in particular: 'Dr Melero was indicating that this was a promising area of his research.' The passage of cross-examination reads as follows:
So there in 2003 or 4,
17 whenever the meeting was, Dr. Melero was indicating that this
18 was a promising area of his research; yes?
19 A. That is what it says, yes.
20 Q. So it would be wrong to say that no one was considering the
21 development of compounds capable of binding the fusogenic
22 intermediate of the F protein?
23 A. Well, I was aware that people were looking at compounds to
24 inhibit fusion. So that would involve the intermediate forms
25 of the F protein.
2 Q. This is in the context of vectored and DNA-based viral
3 vaccines, is it not?
4 A. Yes, I understand. What I am just saying is I knew that
5 people were looking to develop small molecules to inhibit
6 fusion. I was not aware that they were considering it in the
7 terms of a vaccine.
8 Q. Sure. So you were aware that this area of research was there
9 in terms of inhibition, and indeed you had been involved in
10 some of that research, had you not? We saw that on the paper
11 we looked at earlier?
- The Ruiz-Arguello paper is written in the traditional constrained language of scientists who do not, in this type of technical paper, make predictions as to future avenues of research. The issue remains as to who was responsible for that second sentence in the quote from the Boon paper: was it Dr Melero himself, a commentator at the Meeting or one of the authors of the Meeting report? Counsel for GSK considered this an important issue, linked to a later complaint about the identity of the skilled structural biologist. Whether it was Dr Melero or someone else, the fact remains that it was considered worth saying (on the back of the Ruiz-Arguello paper) that 'Future research will be aimed at the development of compounds or antibodies capable of binding to the fusogenic intermediate of the F-protein.' In context, it is likely that this was a comment from Dr Melero or someone in his group because his group were by far the most likely candidates to carry out that future research. In short, therefore, I reject this criticism.
- GSK's other points were that this was not a reference to the prefusion form and in fact teaches away from the prefusion form. I have to address those points below.
- GSK's fourth point concerned a passage of cross-examination on a paragraph from Professor Melero's Chapter in Cane at page 16 in the section headed 'Virus binding and membrane fusion'. The text in question said this:
'Inhibitors of HRSV entry have been the topic of intense research in recent years. Besides the afore-mentioned humanized anti-F monoclonal antibody (Groothuis and Nishida, 2002), small inhibitors of F protein activity have been actively searched. By analogy with synthetic peptides that inhibit human immunodeficiency virus (HIV) replication (Wild et al., 1994), synthetic peptides that reproduce sequences of the heptad repeats (HR) regions of HRSV were synthesized and tested for inhibition of virus infectivity. Peptides containing partial sequences of HRB were found to be highly active inhibitors of HRSV infectivity (Lambert et al., 1996). These peptides presumably bind to the HRA core of an F protein intermediate during the process of virus—cell membrane fusion, blocking latter stages of the fusion process.'
- Counsel put to Dr Taylor that Dr Melero was discussing, in the context of inhibitors, targeting the prefusion form of the F protein, and Dr Taylor agreed. In their written Closing Submissions, GSK labelled this incident as 'The Misreading of Cane'. GSK's point was this passage was talking about an intermediate and not the prefusion form.
- Dr Johnson was cross-examined about the preceding paragraph (which gave reasons why the mechanisms operating during the initial stages of the HRSV replicative cycle may differ to some extent from the mechanisms that operate for entry of other paramyxoviruses) and, subject to a slight qualification, she agreed it represented the CGK. Dr Johnson did not give any evidence about this particular paragraph (and I will be corrected if I am wrong about this). The following paragraph also discusses inhibition of membrane fusion. It is relevant to note, however, the concluding sentence of this section of the Chapter: 'At any rate, inhibition of the F protein activity by synthetic drugs seems a feasible approach for the development of anti-HRSV compounds that may find some future application in the clinic.'
- In my judgment, this criticism was considerably overblown. Whilst I accept the passage in question does not appear to refer to the prefusion form but to an intermediate, it remains a point of relevance. It demonstrates, along with other passages, that attention was being paid in the art to mechanisms and vaccines which would prevent the formation of the postfusion form and the replication process.
- GSK's third main criticism was that Counsel for Pfizer appeared to confuse antibody therapy with vaccination, although no examples were provided.
- It was plain that Calder 2000 used mouse monoclonal antibodies to demonstrate binding to both the prefusion and postfusion forms of the F antigen. It was also plain that the humanised commercial antibody therapy, palivizumab, was likewise understood to bind to both the pre and post-fusion forms of the F antigen. This was one of Dr Taylor's major points. Not surprisingly, these antibody therapies were frequently mentioned in the course of cross-examination. Dr Taylor herself often referred to palivizumab and its ability to bind to sites II and IV, as disclosed in the Calder paper in 2000. So, GSK's submission that 'Antibody therapy has nothing to do with vaccinating humans and has nothing to do with the Patent' was, in my view, significantly overstated.
- Overall, I do not consider this criticism had any merit at all. Throughout the cross-examinations of Drs Johnson and Taylor, there was occasional reference to palivizumab but there did not appear to me to be any confusion.
- GSK's fourth criticism, said to be 'the most troubling' was 'the recasting of the Skilled Team'. GSK contend that this disoriented Dr Taylor and went 'some way' to explaining her corrections. The essence of this complaint was the terminology used in the cross-examination of Dr Taylor to characterise the person in the Skilled Team giving input on structural matters. Where the complaint ended up was that it was said that Dr Taylor was cross-examined on the basis that the Skilled Team included Dr Melero, a world-class structural virologist, a far cry from the structural biologist of ordinary skill in the art.
- GSK devoted 6 ½ pages of their written closing to a close textual analysis of the expressions used in cross-examination. In this section of their closing, GSK mix together a number of issues and significantly overcomplicate what is essentially a simple point.
- By the priority date, Fields was more up to date than Cane, in that Fields referred to (and used illustrations from) both Yin 2005 and Yin 2006, whereas the relevant parts of Cane only referred to Yin 2005 and not Yin 2006. It was clear from the relevant parts of Fields (based on the two Yin papers) that:
i) the PIV5 prefusion F and hPIV3 postfusion F structures 'are in strikingly different conformations';
ii) those conformations were presented in Fields as representative of the paramyxovirus fusion protein;
iii) it was to be expected that RSV, as a related paramyxovirus to PIV, would exhibit the same strikingly different conformations between pre- and postfusion structures.
- Against this backdrop, whether one uses the more general term of 'structural biologist' or the more specific and possibly more apt 'structural virologist', the Skilled Team requires a person with the expertise to understand, model and if appropriate, exploit these structural differences. That, in my view, is what Counsel for Pfizer was getting at in his cross-examination. At certain points, it is true that he used Dr Melero as the exemplar of this expertise. GSK roundly criticised the reliance on Dr Melero on the basis he was a world-class scientist and way above the person of ordinary skill in the art. However, I very much doubt that Dr Taylor thought that she was being invited to combine a vaccinologist of ordinary skill with a structural virologist of exceptional skill.
- I have to return to consider the impact of this point on the allegations of obviousness. At this point I indicate this criticism was again considerably overstated.
Dr Taylor's 'corrections'
- I can now turn to the vexed issue of Dr Taylor's corrections. These occurred in the following circumstances.
- To a significant extent, Dr Taylor's narrow approach in her written evidence was remedied by answers she gave during the first day of her cross-examination, when she agreed that a series of passages in the CGK textbooks represented the CGK. Then, as is customary, she was given the transcript to review overnight so that she could correct any misunderstandings or misstatements in the answers she had given.
- At the start of her second day of cross-examination, Dr Taylor then proceeded to give a number of 'corrections' to answers given on the first day. Her explanation of these 'corrections' lasted for a good 30 minutes. The reason why I put 'corrections' in inverted commas is because they went far beyond corrections in any normal sense of the word.
- Expert witnesses are on oath (or affirmation) and owe duties to the court. If they believe an answer they have given was incorrect, they are permitted, one might say obliged, to correct it. The process of giving expert evidence in court is a tiring one and I do have sympathy for both witnesses and cross-examiners who participate in the process. Naturally, there will be occasional errors, and it is right they should be corrected.
- It is important however, that the ability to suggest corrections to the transcript is not abused. It is inappropriate for a witness to come back the next day and put forward replacement answers which they wish they, perhaps, might have come up with, rather than the evidence they gave in response to clear lines of questioning, which reflected their opinion at the time.
- In Dr Taylor's case there are additional potential factors which I must consider. One is GSK's suggestion that she was getting tired to such an extent that she was unable to concentrate properly on the question put to her. A countervailing point is that she was, in the cross-examination in question, largely being asked about whether passages in the CGK textbooks represented the CGK. In other words, these ought not to have been particularly demanding points.
- Counsel for Pfizer invited me to reject all her 'corrections'. Initially that appeared to be an attractive submission. However, it remained necessary to review each of her 'corrections' in detail, not least because in their written closing, GSK sought to defend her corrections on the basis that her cross-examination on the first day had been unfair.
- In their closing submissions, GSK highlighted certain passages of cross-examination in particular which they submitted demonstrated that Dr Taylor had, in effect, been unable to concentrate on the question. In particular, three of the points I dealt with above, namely at [318]-[320], [321]-[326], [327]-[330], were identified as examples and these related to incidents which occurred during the last session of Dr Taylor's first day of cross-examination. GSK submit that in this session it was evident that Dr Taylor was 'very tired'. Whilst it was a long day which started at 10am, I did not detect at the time that in that session Dr Taylor was unable to concentrate on the questions being put to her, although, on a few occasions, I intervened to ensure that the witness had located and read the passage the subject of Counsel's impending question. In any event, if a witness does appear to be losing the ability to concentrate, his or her legal team is far more likely to be able to detect those signs than a Judge who is encountering the witness for the first time. The onus should be on the legal team to speak up, particularly where it is the first time the witness has given expert evidence in a patent case.
- The first was the incident which I discussed in paragraphs [318]-[320 above]. I do not consider this supports GSK's contention at all. Neither does the point I discussed at [321]-[326]. The point discussed at [327]-[330], however, does.
- For that reason I have had to give careful consideration (a) to Dr Taylor's corrections, (b) the context of the answers she sought to correct and (c) the cross-examination overall.
- It is not necessary to discuss every one of Dr Taylor's corrections but I will address some further examples (which appeared to me to be the most significant).
- In a number of her 'corrections' Dr Taylor made explicit or implicit reference to the Calder 2000 paper.
- To put this example into context, Dr Taylor was being asked questions about Chapter 41 of Fields, the Chapter on Paramyxoviridae. This Chapter was bang up to date, making reference to Yin 2005 (fn 421) and Yin 2006 (fn 422) and featuring Figures adapted from those papers showing, in Fig 41.15 the F Protein prefusion conformation and, in Fig 41.16, the F protein postfusion conformation.
- Her attention was directed to the following passages from Chapter 41 of Fields and in each case she agreed that what was stated was 'known', in the sense of being CGK. This was hardly surprising bearing in mind she agreed that Fields was a source of CGK and this was obviously a centrally relevant chapter.
- Under the heading Paramyxovirus Fusion Protein, the F proteins are described:
The F proteins are homotrimers (55,319,421,422) that are synthesized as inactive precursors (FO). To be biologically active, they must be cleaved by a host cell protease at the cleavage activation site. Cleavage releases the new N-terminus of F1, thus forming the biologically active protein consisting of the disulfide-linked chains F1 and F2 (159,331). The paramyxovirus F genes encode 540 to 580 residues (Fig. 41.11). The F proteins are type I integral membrane proteins that span the membrane once...
The F protein is believed to drive membrane fusion by coupling irreversible protein refolding to membrane juxtaposition and by initially folding into a metastable form that subsequently undergoes discrete/stepwise conformational changes to a lower energy state (176, 203). The F protein found on virions is considered to be in a prefusion form, and after membrane fusion has occurred, the F protein is considered to be in a postfusion form. Cleavage of FO primes the protein for membrane fusion. The varying nature of the residues found at the cleavage site, the enzymes involved in cleavage, and the role of cleavage in pathogenesis is discussed later in this chapter.
- Over the page, under the heading 'Atomic Structures of the Paramyxovirus F Protein' there are subheadings for the Prefusion and Postfusion forms. Under that first subheading:
The atomic structure of the PN5 F protein in its uncleaved metastable prefusion form has been determined (422). To solve the atomic structure, the secreted F protein was stabilized by the addition of a soluble trimeric TM domain (GCNt) that supplants the hydrophobic TM domain.
- Under the second sub-heading 'Structure of the Postfusion Form of the F Protein', the text reads:
The atomic structure of intact uncleaved F protein in its postfusion form (421) has also been determined.
- Under the next sub-heading 'Comparison of the Pre- and Postfusion F Structures':
The PIV5 prefusion F and hPIV3 postfusion F structures are in strikingly different conformations (Fig. 41.17), consistent with a transition from pre- to postfusion forms. None of the intersubunit contacts are conserved in the pre- and postfusion forms. The two F structures are related by flipping the stalk and TM domains relative to the F head. Substantial compacting of the head is observed in hPIV3 postfusion F compared to PN5 prefusion F. DI domains pivot slightly inward, shearing intersubunit contacts, and DII domains swing across, contacting neighboring subunits. Individual DI and DII domains in the two structures remain similar. Potentially related forms of the F protein have been observed in electron micrographs of RSV F (41,124,315,316)
- Having gone through those (and other passages of less central relevance), Dr Taylor was then directed to Figures 41.15 and 41.16 (which depict the PIV5 F protein prefusion and postfusion structures respectively):
20 Turn back now, and just bearing what we have read there
21 in mind and looking at these two pictures, 41.15 and 41.16, what
22 the skilled team would learn from this was that this change in
23 structure between the pre and postfusion forms that had been
24 observed in PIV appeared likely to be reproduced and be part
25 of RSV F. That is correct, is it not?
2 A. Yes.
3 Q. Where it was said that the contact between no subunits were
4 conserved, a consequence of that would be that it would be
5 thought likely that conformational epitopes would change
6 between the pre and postfusion forms, would it not?
7 A. It could be expected to be, yes.
8 Q. Yes. Look at this way. It is possible that it would not, but
9 with such a dramatic conformational change, the expectation
10 would be that conformational epitopes would be disrupted,
11 correct?
12 A. Yes.
13 Q. Can I just turn to 1473? Sorry, 1472, I beg your pardon. At
14 1472, it says: "Cleavage Activation. As discussed
15 previously, the precursor F0 molecule", this is on the
16 right-hand side of 1472, "is biologically inactive and
17 cleavage of the F0 to the disulfide linked chains F1 and F2
18 activates the protein, rendering the molecule fusion-active
19 and permitting viral infectivity." That would have been known
20 as an approach?
21 A. Yes.
- All these answers were part of the flow of the cross-examination. However, the first two answers I have put in italics were 'corrected' by Dr Taylor to read as follows:
i) Line 7: 'It is possible.'
ii) Line 12: 'Not necessarily, because the Calder paper suggests that conformational epitopes in antigenic areas 2 and 4 are not disrupted.'
iii) As for the answer at Line 21, she said 'I said 'Yes' but I was not clear what the approach to what was at that stage.'
- The cross-examination then turned to Dr Melero's Chapter in Cane (Chapter 1, entitled 'Molecular Biology of Human Respiratory Syncytial Virus'). This chapter is not as up to date as the discussion in Fields, referencing only Yin 2005 and not Yin 2006. Nonetheless, Fig 5 'Scheme of the F protein of hRSV' has a rubric and a 3-D colour model derived from Yin 2005. Part of the discussion of the F glycoprotein is as follows:
'The 3-D structure of HRSV F has not been solved to date. However, based on a partial atomic structure of the Newcastle Disease Virus (NDV) F protein (Chen et al., 2001), a partial model of HRSV F was proposed (Smith et al., 2002). A more complete model was later constructed by grafting the six-helix core of HRSV F (Zhao et al., 2000) onto the model based on the NDV F structure (Morton et al., 2003). Fig. 5 shows a similar model of the HRSV F 3-D structure, based on the atomic structure of a soluble form of the parainfluenza virus type 3 F protein recently determined by X-ray crystallography (Yin et al., 2005). The 3-D models of HRSV F are in good agreement with EM images of HRSV F molecules (Calder et al., 2000). Furthermore, when the residues altered in monoclonal antibody escape mutants are located in the F protein model, a good correlation is observed between the location of those residues in the 3-D model and the binding sites of the corresponding antibodies, as observed by EM (Calder et al., 2000) (Fig. 5).
Although these data lend support to the 3-D model of HRSV F, it is still not known whether the structure presented in Fig. 5 corresponds to the conformation adopted by the F molecule in the pre- or postactive configuration (see later).
Interestingly, highly neutralizing antibodies directed against the F protein bind to epitopes at antigenic sites II or the overlapping sites IV, V and VI, while other antibodies specific to these sites are only weakly neutralizing. Monoclonal anti-bodies binding at epitopes of site I have all low neutralizing activity (Beeler and van Wyke Coeling, 1989; Garcia-Barreno et al., 1989; Arbiza et al., 1992; Lopez et al., 1998). Thus, in contrast to the aforementioned neutralization by anti-G antibodies based on steric hindrance, the mechanism of HRSV neutralization by anti-F anti-bodies seems to require specific interactions of antibodies with certain residues of the F protein, perhaps to inhibit conformational changes that occur during the process of membrane fusion. The importance of research into anti-F antibodies is emphasized by the fact that a humanized neutralizing monoclonal antibody (Palivizumab), directed against an epitope of F protein antigenic site II, is the only product available to date for prophylactic treatment of HRSV infections in high-risk infants (Groothuis and Nishida, 2002)."
- I have underlined a key sentence in this passage for two reasons. First, because, when Counsel for GSK cross-examined Dr Johnson on this passage, he skipped over this sentence. Second, because it was basis of and indeed the 'central passage' in this passage of cross-examination of Dr Taylor:
"Can
19 I put it this way. I think your answer suggested that it was
20 an area of interest, but it was not certain, there was not
21 anything saying "this is the place you must go for prefusion";
22 is that fair?
23 A. That is correct, yes.
24 Q. So can I suggest this; the skilled team would have been
25 fertile ground for suggestion that the prefusion form was one
to go for, it would not have seemed an outrageous or an
3 outlandish suggestion to them at all at the priority date,
4 would it?
5 A. No. It depends how unimaginative the person is supposed to
6 be.
7 Q. Your concern with this state of CGK was that there was
8 speculation that it might be that the prefusion form was
9 interesting, and was a useful target, but it had not been
10 determined. That is correct, is it not?
11 A. It had not been determined, and there was evidence that there
12 was not major antigenic differences, as I referred to before
13 in the Calder paper, neutralising antibodies to sites II and
14 IV addressed here, bound to both cones and lollipops in
15 electron microscopy.
16 Q. What we get in this central passage here, and would have been
17 in the minds of the skilled team, is a suggestion that
18 neutralisation by anti-F antibodies seems to require specific
19 interactions with certain residues of the F protein, perhaps
20 to inhibit conformational changes, and so although there might
21 well be, when one looks at the Calder paper, neutralising
22 antibodies that bind both forms that were observed, the
23 skilled person would be very interested in the thought of
24 neutralising by antibodies that bound the prefusion form;
25 correct?
2 A. Quite possibly, yes. Not unreasonable.
3 Q. Not unreasonable. If you have two targets, pre- and
4 postfusion, you would certainly think well, the prefusion is
5 an obvious way to go; yes?
6 A. Yes, okay.
7 Q. As at the priority date?
8 A. Yes."
- Again, I put the answers which Dr Taylor sought to 'correct' in italics. She explained her corrections in this passage, starting with the answer at line 2:
"I would like to change that, again, to "Not
6 necessarily", because monoclonal antibodies to antigenic area
7 sites 2 and 4 are already known to bind to both forms, and
8 that is Calder again. Then in my answer at line 6, "the
9 prefusion is an obvious way to go", I would like to delete
10 "Yes, okay." "There is no common general knowledge that PreF
11 was being proposed as a subunit vaccine at that time."
- In these and other 'corrections', there was such frequent reference to Calder 2000 that at the end of her series of corrections, I was moved to ask Dr Taylor whether it was her position that the art did not really move on between Calder 2000 and the priority date. Her answer seemed to relate to the content of the Calder paper, followed by 'I am not aware of anything else in that area'.
- Counsel for Pfizer put to her that her corrections were a sort of 'l'esprit de l'escalier' which he explained to the witness along the lines of 'answers she had come up with afterwards to support an argument which she did not give at the time'. Her response was that 'I was getting a bit confused at times, because of the line of questioning and tiredness, and I just wanted to make sure the record was correct.'
- I am acutely conscious that the answers which Dr Taylor sought to correct were given either side of a mid-afternoon break (at 3.10pm), that Dr Taylor endured a long day of cross-examination which had commenced at 10 a.m. and that Counsel for GSK sought to defend her corrections on the basis that she was very tired when she gave the answers she sought to correct the following day.
- Furthermore, as I explained above, I identified one of her corrections which appeared to me to be legitimate. This related to what GSK labelled 'the Misreading of Cane', as identified above at [328]. On reflection, it seems to me that Dr Taylor's correction here was correct. The passage does seem to be addressing possible intermediates. At the very least, it is certainly not clear that this passage is talking about the prefusion form. So the fact that Dr Taylor simply answered 'Yes' indicates she may well have been getting tired and losing the ability to concentrate on these intense technical questions. It is for this reason that I have reviewed all the 'corrections' with special care. Having done so, this single correction was very much the exception. With that exception, almost all of her corrections fell into one of the following categories. Either they were not an answer to the question which had been posed and/or the corrected answer was completely at variance with the flow of the cross-examination and/or the corrected answer completely ignored the statement put to her from a CGK textbook.
- I have reviewed these sections of the transcript in detail but I am driven to the conclusion that, in making these corrections, Dr Taylor was arguing GSK's case and closing her mind to the plain sense of the textbooks. Indeed, these corrections cast very considerable doubt on the independence and objectivity of all of her evidence unless corroborated by contemporaneous material. A further consequence is that GSK's attempts to defend her corrections were largely an exercise in misdirection.
- As I have already indicated, a lot of energy and cross-examination was directed by GSK to two particular paragraphs in Dr Johnson's first report, which were flagged as providing the foundation for her opinions that the Patents were obvious over the cited prior art. I observe in passing that this gave the impression at times that GSK rather wished they were facing purely an obviousness attack over the CGK. The two paragraphs in question are [124] and [125] of her first report in which Dr Johnson said the CGK as to the PreF and PostF conformations of RSV went some way beyond what I recorded as agreed in [160] above. To aid analysis, I have labelled and underlined the individual sentences and highlighted particular terms which were the subject of challenge by GSK:
124. (A) It was widely accepted that effective neutralizing antibodies should bind the native conformation on the virion. (B) Furthermore, the RSV Vaccinologist would have known that it had been demonstrated that "mature" forms of RSV F as found on the virion surface could elicit highly neutralizing antibodies, whereas other forms did not. It was also thought that the FI-RSV vaccine (mentioned above and discussed further below) only induced low levels of neutralizing antibodies because antigenic epitopes had been altered by the process of formalin-inactivation. (C) By the Priority Date, it was suspected that the F protein in FI-RSV could have been in a postfusion conformation.
125. (D) The RSV Vaccinologist would have known that it was thought that the most effective neutralizing antibodies would likely bind to the prefusion conformation of the F protein. This was based on the principle that antibodies that bound to the prefusion form would prevent transition to the postfusion state and block fusion of the membranes, thereby blocking virus infection. This hypothesis was well established for certain other fusion proteins such as from HIV-1 and influenza.
- These points engage a number of the CGK disputes which I identified in [82] above. Before dealing with the labelled passages, I should record some general points which arose in cross-examination. Dr Johnson was asked about how she had prepared the CGK section of her report. She said that she had written the CGK section of her report 'primarily' from memory. When pressed as to how she was able to date the state of knowledge to 2007, she explained that after she had prepared the initial section, a bit later we (i.e. the solicitors and her) 'went through and put in all the references' and that 'the primary references throughout have always been Fields first and then Cane, as the sources of common general knowledge.' As Counsel pointed out, the references do not appear in her report but she explained that they were in a 'middle version', and later she was asked to take the references out before her first report was served.
- Having gone to the trouble of inserting references, I am bound to say it would have been far more helpful if they had been retained in the report as served but Dr Johnson cannot be blamed for this because she did what the solicitors asked of her. The insertion and then the removal of these references gives rise to a suspicion that the possible absence of references against certain propositions might have served to highlight that their status as CGK was questionable.
- Another 'anchor' which enabled Dr Johnson to date to 2007 was her filing of a patent application. The provisional application was filed on 30 November 2006 and the PCT on 30 November 2007. Counsel cross-examined on the content of this patent application US2010/0247621 A1 but all that seemed to establish was that neither the application nor the work leading up to it was relevant to the issues I have to decide.
- Later in her cross-examination a similar point arose. In her first report, Dr Taylor said she had no recollection of having discussed the idea of vaccinating with the F protein in its prefusion conformation. Dr Johnson responded in her reply report saying she recalled being involved in discussions about the importance of generating antibodies to the native prefusion conformation. As an example, she cited a discussion with participants at the 2003 RSV Symposium at Stone Mountain in Georgia. In cross-examination she gave further examples and details. She said there were public discussions at some of the meetings, after Dr Melero's presentations and, specifically in respect of the 2003 RSV Symposium, that a (brief) discussion took place by the pool, it involved Dr Barney Graham, Dr Melero, Ed Walsh, Larry Anderson who were sitting by the pool, discussing the lollipops and antibodies. Drs Johnson and Tripp were walking past and briefly engaged in the conversation, bringing up the G protein since that was the primary focus of their research. Counsel then suggested the discussion was only about antibodies binding to G, but Dr Johnson's recollection was they were talking about antibodies in general binding to the lollipops.
- Counsel for GSK attempted to dismiss this evidence on the basis that it was only about the G protein and not F, that is not correct. In my view, Dr Johnson had a good recollection of this incident.
- Dr Johnson was challenged strongly on her evidence that, having read Yin and attended the Jardetzky presentation, she considered at the time that a prefusion subunit vaccine was the way to go. She explained she did not choose to follow that line for several interconnected reasons: first, so far as the F protein was concerned, she was following a vector-based approach using the full length wild-type F expressed in 15 or 17 vector systems, an approach which did not involve or need stabilisation; second, due to the skill sets available in her lab, the inference being that they did not have access in their lab to a structural biologist (at least until Jason McLellan had a seat in their lab in 2010).
- The removal of supporting references was particularly significant for these central paragraphs [124] & [125].
- Read out of context, [124] may appear to be a curious mix of points about effective neutralizing antibodies (points A & B) with points about FI-RSV (in the third sentence and point C). However, those two sentences arose from some earlier paragraphs in her report. There was not much focus on the third sentence, but I can deal with point C here. The cross-examination resulted in Dr Johnson agreeing to put a line through point C. GSK submitted that this was indicative of the lack of care with which Dr Johnson prepared her evidence on CGK, but the force of this submission can only be assessed in the round.
- The remaining points - A, B & D are related. The first point to address is the debate during the cross-examination of Dr Johnson over what was meant in the art by the 'native' or 'natural' conformation and the 'mature' form on the virion.
- It was clear from Dr Johnson's evidence that she considered that references to the "natural", "mature", or "native" form of RSV F were generally understood by the RSV Vaccinologist to be the metastable prefusion conformation. GSK did not accept this interpretation and submitted that that whereas these terms were used to describe the antigen on the virus they were never used to distinguish the pre and postfusion forms of the F antigen.
- Pfizer's position was that whilst the Patents define the term "native" and "naturally occurring" to mean proteins or polypeptides that are present in the same state as they are in nature, i.e. not modified artificially, such as different naturally occurring strains of RSV ([0042]), the term "native" was also used widely at the Priority Date to refer to the prefusion form of the F protein present on the surface of cells. GSK's position was that it is incorrect to say that "natural" excludes the postfusion form.
- Much of the cross-examination appeared to be an issue of linguistics, the key issue is whether RSV Vaccinologists were considering the different conformations of the RSV F protein in their approach to vaccine design. In light of the conclusions above very little therefore turns on this particular point. For example, Calder 2000 referred to the preactivated structure of F and the postactivation structure.
- This debate showed that there was a difference in terminology between vaccinologists and structural biologists/virologists. Dr Johnson had adverted to this in her third report where she said that the term 'prefusion' (as opposed to natural or native) had not been specifically used in the RSV vaccine literature at the priority date. Vaccinologists had tended to refer to the native or mature conformation, whereas structural biologists called that the prefusion conformation. It is true that in the cross-examination of Dr Johnson, Counsel produced examples of papers where the term 'native' was used in particular contexts to refer to both the prefusion and postfusion forms, but in general I was satisfied that Dr Johnson was correct in referring to the 'native' conformation as equivalent to the prefusion form. Furthermore, that usage was amply reinforced by Professor Weissenhorn's evidence. He was plainly using prefusion and native as alternatives and indicating this was generally accepted usage by structural biologists. Take this example (which I accept was CGK to the Skilled Structural Biologist):
'It would also be CGK that as a matter of first principles an antigen which better mimics the native, prefusion form of the F protein (present on infectious virions) would be more likely to generate antibodies that interfere with cell entry and so could serve as better immunogens. This concept was well-known in the HIV field. Indeed, by the Priority Date, a number of stabilization strategies had been used to try to preserve the native trimeric conformation of the HIV-1 Env protein. These approaches were summarised in a number of review articles available at the time, including for example articles by Burton 2002 (citing "Neutralizing antibody (*) binds to native envelope spikes.."), Burton 2006 (review of different Env stabilization approaches to generate native Env antigens), Haynes and Montefiori 2006 (citing "the lack of current immunogens that mirror the native envelope structures needed to induce neutralizing antibodies .."), Nabel 2002 (citing "..expression in a conformation that more closely resembles that of the native protein..").'
- In her third report and in cross-examination Dr Johnson did identify references from Fields, Cane and other papers which supported the notion it was CGK that the prefusion/native/mature form on the virion could elicit highly neutralizing antibodies. She was also confident to mention other textbooks in cross-examination which would mention this 'basic principle'. It suffices to set out the following:
i) First, at p1466-7 in Fields, Chapter 41 (Paramyxoviridae: Their Viruses and Their Replication)::
"The F protein is believed to drive membrane fusion by coupling irreversible protein refolding to membrane juxtaposition and by initially folding into a metastable form that subsequently undergoes discrete/stepwise conformational changes to a lower energy state (176,203). The F protein found on virions is considered to be in a prefusion form, and after membrane fusion has occurred, the F protein is considered to be in a postfusion form."
ii) Second, in Cane at p50, under the heading 'Antibody recognition of RSV proteins' the paragraph concludes with this:
'Analysis of the repertoire of F-specific antibodies induced by RSV infection demonstrated that highly neutralising antibodies recognised the mature F protein on the cell surface and on virions, whereas poorly neutralising antibodies appeared to recognise immature F protein (Sakurai et al., 1999).'
iii) Third, at p488 in Fields, Chapter 15 (Immunization against viral diseases):
"First, antiviral Abs against the extracellular domain of surface proteins predominantly recognize conformational epitopes. Such conformational epitopes are difficult to mimic with peptides or other forms of an immunogen in which the surface protein antigenic sites are denatured. An immunogen that possesses the structures of the native protein most effectively induces Abs that recognize the conformational epitopes that mediate immunity."
- Dr Johnson cited that last passage as an example illustrating '[t]he importance of the native form of the F protein as expressed on the surface of virions in generating fusion-inhibiting, neutralizing, and protective antibodies would have been part of the CGK of the RSV Vaccinologist.' Indeed, Fields gives that passage as the first of a number of generalisations which can be made about the nature of antibody and its interaction with virus surface proteins.
- Furthermore, the passage I cited above from Professor Weissenhorn's first report (written before any issue arose) not only supported Dr Johnson's use of 'native' to represent the prefusion form, but also the notion that the native or prefusion form as a target for inducing neutralizing antibodies was under discussion.
- In the light of the considerations I have discussed above, I am inclined to find that points A, B and D from [124] and [125] of Dr Johnson's first report were CGK. This conclusion must remain an interim one until after I have considered the impact of GSK's case on secondary evidence. This is because of GSK's remaining argument that these important facets of CGK could only be assembled with the benefit of hindsight and they were not apparent to persons of ordinary skill in the art at the priority date.
- I can now revert to consider the CGK disputes which I listed at [82] above. A number of those disputes overlap with points A, B & D and conclusions on those also remain interim.
i) The extent to which adjuvants were necessary or generally used or expected to be necessary in subunit vaccines.
- Although adjuvants have been discussed above, this is a particular issue which remains to be decided and it is relevant to Pfizer's insufficiency plea.
- Pfizer's case based on the written evidence was that subunit vaccines consisting of recombinant proteins were known to be generally poor immunogens when administered without an adjuvant.
- During cross-examination, Dr Taylor made clear that she was not aware of a subunit vaccine for any other disease that has been approved without an adjuvant. Similarly, as at the priority date, Dr Johnson was not aware of any protein-based vaccine that did not use an adjuvant. As the evidence pointed to the extreme difficulties in making a protective immune response for a protein-based vaccine without an adjuvant, I agree that the presence of an adjuvant would have generally been considered as necessary.
ii) Whether RSV Vaccinologists were considering the different conformations of the RSV F protein in their approach to vaccine design.
- Pfizer's case was that it was understood at the priority date that the F protein existed in a metastable prefusion conformation and a thermodynamically stable post fusion conformation. Their position is that this would lead to the consequence that (i) the prefusion conformation was of interest generally to the skilled team and (ii) for the skilled team to investigate the RSV F prefusion conformation, it would be desirable to stabilise it in some way. GSK's case is that, in reliance on Dr Taylor's recollection and a sample of 25 review papers from 2000-2008, RSV Vaccinologists were not considering these different conformations of the RSV F protein in their approach to vaccine design. GSK submitted that her recollection is consistent with the literature.
- It may be that the review papers made no mention of the prefusion and postfusion conformations, but the textbooks indicate otherwise. In my view, the evidence indicates at the Priority Date it was understood that the F glycoprotein existed in a metastable prefusion conformation and a thermodynamically stable postfusion conformation. In nature the F protein is found on virion on the membrane and is considered to be in a prefusion form. After membrane fusion has occurred the F protein is considered to be in a postfusion form.
- This understanding was based on the general mechanism for class I viral fusion proteins known by the skilled team and discussed (at the least) in the textbooks, such as Fields and Cane.
- From the evidence it appears that the skilled team at the priority date understood that the general mechanism for class I viral fusion proteins posits the folding of the uncleaved protein to a metastable state, which can be activated to undergo large conformational changes to a more stable fusogenic or postfusion state. The attainment of the prefusion conformation, its regulation and relative free energy as compared to the postfusion form are all key to the process by which class I viral fusion proteins function.
iii) The relevance of researching recombinant subunit vaccines for related viruses to RSV which share the Class I fusion protein mechanism known to be a main target for RSV vaccines, including other paramyxoviruses, and HIV-1 and influenza HA.
- Pfizer relies on analogy with other viruses such as HIV and influenza, with Dr Johnson providing evidence that the skilled vaccinologist would have followed developments in other paramyxoviruses and major developments in other viruses such as HIV-1, influenza A virus, and Ebola virus.
- GSK's position was that even if the skilled vaccinologist did have regard to the major developments in vaccine design from the influenza, parainfluenza or HIV fields, there is nothing in that body of CGK which would guide an approach towards the prefusion form for an RSV F subunit. GSK further submitted that in any event, that the RSV F protein is metastable and switches to postfusion when solubilised, whilst influenza HA has the brakes already applied and the major switch is the lowering of pH. Further, influenza enters the target cell in a different way to RSV. GSK further argued that with respect to HIV that in light of the extensive publications that the skilled vaccinologist working on RSV would not have kept up with such developments.
- In her cross-examination, Dr Taylor agreed that in terms of the operation, the pathology, the infective pathways, the structural analysis of RSV, PIV and the other paramyxoviridae, there was cross-fertilization between them. She agreed that a skilled team interested in RSV would be interested in what was going on and what was being learned from PIV and vice versa. This position was also confirmed by Dr Johnson.
- Fields indicates that the team would not consider RSV F in isolation. In the analogies being drawn between PIV and NDV and the whole family it would be clear that information that was learnt about, say the PIV structure, would be relevant to the investigation of RSV F.
- The F subunit of RSV and PIV in particular were viewed as closely analogous at the priority date. The F protein is highly conserved to other type I, class I viral fusion proteins at the structural and functional level, both within the paramyxovirus family and more broadly in class I fusion proteins such as HIV-1 and influenza HA. The skilled person would have known that the paramyxovirus F proteins belong to the class I viral fusion protein type, the longest standing member of which is the influenza virus HA.
- Furthermore, Professor Weissenhorn's evidence provided powerful support for the cross-fertilisation amongst the class 1 fusion proteins, as his evidence on the CGK demonstrated.
- To summarise therefore, I find that this would have been known to and considered by the Skilled Team.
iv) Whether references in the papers to the "natural", "mature" or "native" F protein would be understood by the skilled vaccinologist as the "prefusion" form.
- I have already addressed this issue. Although it depends on the context, in general references to the natural, mature or native F protein would be understood by the skilled vaccinologist as the prefusion form.
v) Whether it was known that the most effective neutralizing antibodies would be likely to bind to the prefusion conformation of the F protein.
- This is point D above.
- Pfizer's case was that it was thought that the most effective neutralizing antibodies would be likely to bind to the prefusion conformation of the F protein.
- GSK accepted that it was widely accepted that effective neutralizing antibodies should bind the native conformation on the virion. They noted that it is self-evident that neutralising antibodies will bind to some form of the protein which is found in nature, as opposed to a form which is unnatural such as a denatured protein.
- In light of the finding above that class I fusion proteins would have been relevant to the skilled team, given that it was well established in relation to class I fusion proteins that antibodies that bound to the prefusion form would prevent transition to the postfusion state and block fusion of the membranes, it appears that it was known that it was thought that the most effective neutralizing antibodies would be likely to bind to the prefusion conformation of the F protein.
- A subsidiary point concerns the use of the expression 'most effective'. This emphasises the distinction between binding to prefusion ("most effective") as opposed to binding to postfusion (implicitly "least effective"). GSK say that is a distinction which was not being drawn in the art and was not part of the common general knowledge - pre- and postfusion both being recognised by neutralising antibodies. This seems to be another reference to the Calder argument, which misses the point.
vi) Whether the skilled team had an awareness of and expertise in stabilising fusion glycoproteins, and that this was important for structural studies and for immunization experiments in a vaccine context.
- Pfizer's position is that the skilled team had an awareness of and expertise in stabilising fusion glycoproteins, and that this was important for structural studies and for immunization experiments in a vaccine context. A number of strategies had already been developed and tested in HIV-1 with the aim of preserving the native Env trimer conformation. At the priority date, it was known that F proteins are predominantly in a metastable prefusion conformation on the virion surface, which folds to a stable postfusion conformation.
- Dr Taylor agreed that if you want to study the metastable state then you would have to stabilize it so it became less meta, more stable. If that conformation is what you are interested in, it would be an obviously desirable thing to do to stabilize it in that conformation.
- It will be apparent from what I have already said that, in my view, it is plain that the skilled team had to have expertise in stabilising fusion glycoproteins and that the skilled team was aware that this was important for structural studies and for immunization experiments in a vaccine context.
- What I have set out above is more than enough technical background to understand the Patents. This is a case in which the Patents provide an important indication as to the skills and knowledge of the Skilled Team.
- The parties only found it necessary to address the specification of EP 258, except where indicated, and I will do the same. The specification repays careful reading, for reasons which I explain below. Generally, there is a good deal of information in the specification aimed at the Skilled Structural Biologist, some of the subtleties of which might not be fully understood by the Skilled Vaccinologist.
- The field of the invention is identified in [0001]. The invention is said to relate to "compositions and methods for eliciting an immune response specific for Respiratory Syncytial Virus".
- The Background section identifies RSV as being one of the most common infections in the world. At [0004] it is noted that whilst there have been attempts to produce a safe and effective RSV vaccine that produces durable and protective immune responses, "none of the candidates evaluated to date have been proven safe and effective as a vaccine for the purpose of preventing RSV infection and/or reducing or preventing RSV disease, including lower respiratory infections (LRIs)."
- The Summary section beginning at [0005] notes:
"This disclosure concerns recombinant respiratory syncytial virus (RSV) antigens. More specifically, this disclosure concerns antigens including a recombinant F protein that has been modified to stabilize the trimeric prefusion conformation. The disclosed recombinant antigens exhibit superior immunogenicity, and are particularly favorably employed as components of immunogenic compositions (e.g., vaccines) for protection against RSV infection and/or disease".
- Then at paragraph [0006]:
"Specifically, a recombinant respiratory syncytial virus (RSV) antigen is provided which assembles into a trimer, comprising a soluble F protein polypeptide comprising an F2 domain and an F1 domain of an RSV F protein polypeptide and comprising an amino acid sequence comprising a heterologous trimerization domain positioned C-terminal to the F1 domain that stabilizes the prefusion conformation of the F protein"
- Those paragraphs are different in the EP 710 patent, notably as at [0005]:
"The present invention relates to a recombinant respiratory syncytial virus (RSV) antigen comprising a soluble F protein polypeptide comprising an F2 domain and an F1 domain of an RSV F protein polypeptide, wherein there is no furin cleavage site between the F2 domain and the F1 domain, and wherein the polypeptide further comprises a heterologous trimerization domain positioned C-terminal to the F1 domain."
- Reverting back to EP258, paragraph [0010] explains:
"The present disclosure concerns recombinant respiratory syncytial virus (RSV) antigens that solve problems encountered with RSV antigens previously used in vaccines, and improve the immunological as well as manufacturing properties of the antigen. The recombinant RSV antigens disclosed herein involve a Fusion (F) protein analog that include a soluble F protein polypeptide, which has been modified to stabilize the prefusion conformation of the F protein, that is, the conformation of the mature assembled F protein prior to fusion with the host cell membrane."
- The first sentence in paragraph [0011] introduces Figure 1A and Figure 1B. Figure 1A is the RSV F protein wild-type and a schematic illustration of the exemplary PreF antigens is provided in Figure 1B. These figures are set out below in connection with [0152] which explains them in greater detail. The specification explains:
[0012] With reference to the primary amino acid sequence of the F protein polypeptide (FIG. 1A), the following terms are utilized to describe structural features of the PreF antigens.
[0013] The term F0 refers to a full-length translated F protein precursor. The F0 polypeptide can be subdivided into an F2 domain and an F1 domain separated by an intervening peptide, designated pep27. During maturation, the F0 polypeptide undergoes proteolytic cleavage at two furin sites situated between F2 and F1 and flanking pep27.
- [0014] discusses the subject of the invention.
'The prefusion F (or "PreF") antigen is a soluble (that is, not membrane bound) F protein analog that includes at least one modification that stabilizes the prefusion conformation of the F protein, such that the RSV antigen retains at least one immunodominant epitope of the prefusion conformation of the F protein. The soluble F protein polypeptide includes an F2 domain and an F1 domain of the RSV F protein (but does not include a transmembrane domain of the RSV F protein).'
Stabilization
- The specification then turns to discuss various stabilization approaches:
'[0015] The PreF antigens are stabilized (in the trimeric prefusion conformation) by introducing one or more modifications, such as the addition, deletion or substitution, of one or more amino acids. One such stabilizing modification is the addition of an amino acid sequence comprising a heterologous stabilizing domain. In exemplary embodiments, the heterologous stabilizing domain is a protein multimerization domain. One particularly favorable example of such a protein multimerization domain is a coiled-coil domain, such as an isoleucine zipper domain that promotes trimerization of multiple polypeptides having such a domain. An exemplary isoleucine zipper domain is depicted in SEQ ID NO:11. Typically, the heterologous stabilizing domain is positioned C-terminal to the F1 domain.'
- Following a reference to a possible linker in [0016], [0017] describes an alternative stabilisation approach:
'[0017] Another stabilizing modification is the elimination of a furin recognition and cleavage site that is located between the F2 and F1 domains in the native F0 protein. One or both furin recognition sites, located at positions 105-109 and at positions 133-136 can be eliminated by deleting or substituting one or more amino acid of the furin recognition sites, such that the protease is incapable of cleaving the PreF polypeptide into its constituent domains. Optionally, the intervening pep27 peptide can also be removed or substituted, e.g., by a linker peptide. Additionally, or optionally, a non-furin cleavage site (e.g., a metalloproteinase site at positions 112-113) in proximity to the fusion peptide can be removed or substituted.'
- At [0018], another example of a stabilizing mutation is described, with examples. This becomes relevant when we reach Example 1 (emphasis added):
'[0018] Another example of a stabilizing mutation is the addition or substitution of a hydrophilic amino acid into a hydrophobic domain of the F protein. Typically, a charged amino acid, such as lysine, will be added or substituted for a neutral residue, such as leucine, in the hydrophobic region. For example, a hydrophilic amino acid can be added to, or substituted for, a hydrophobic or neutral amino acid within the HRB coiled-coil domain of the F protein extracellular domain. By way of example, a charged amino acid residue, such as lysine, can be substituted for the leucine present at position 512 of the F protein. Alternatively, or in addition, a hydrophilic amino acid can be added to, or substituted for, a hydrophobic or neutral amino acid within the HRA domain of the F protein. For example, one or more charged amino acids, such as lysine, can be inserted at or near position 105-106 (e.g., following the amino acid corresponding to residue 105 of reference SEQ ID NO:2, such as between amino acids 105 and 106) of the PreF antigen). Optionally, hydrophilic amino acids can be added or substituted in both the HRA and HRB domains. Alternatively, one or more hydrophobic residues can be deleted, so long as the overall conformation of the PreF antigen is not adversely impacted.'
- At [0019] it is explained that any and/or all of the stabilizing modifications can be used individually or in combination and
'In exemplary embodiments the PreF protein comprising a polypeptide comprising an F2 domain and an F1 domain with no intervening furin cleavage site between the F2 domain and the F1 domain, and with a heterologous stabilizing domain (e.g., trimerization domain) positioned C-terminal to the F1 domain. In certain embodiments, the PreF antigen also includes one or more addition and/or substitution of a hydrophilic residue into a hydrophobic HRA and/or HRB domain. Optionally, the PreF antigen has a modification of at least one non-furin cleavage site, such as a metalloproteinase site.'
- As an aside, I note that Dr Taylor described the modifications referred to in [0015]-[0019] as molecular biology techniques.
- At paragraph [0023] on page 6, there is an introduction of Figure 10. In EP258, the construct that is of importance is the sMP340-A construct:
"Fig. 10 depicts cartoons of the mature RSV protein" "and the three RSV soluble fusion (sF) protein constructs ... used in our studies."
Immunogenic compositions
- [0024]-[0025] discuss the use of the disclosed PreF antigens in immunogenic compositions to be favourably deployed as vaccines for RSV:
'[0024] To enhance the immune response produced following administration, the immunogenic composition typically also includes an adjuvant. In the case of immunogenic compositions for eliciting a protective immune response against RSV (e.g., vaccines), the compositions favorably include an adjuvant that predominantly elicits a Th1 immune response (a Th1 biasing adjuvant).
[0025] The immunogenic compositions described herein are favorably employed as vaccines for the reduction or prevention of infection with RSV, without inducing a pathological response (such as vaccine enhanced viral disease) following administration or exposure to RSV.'
- Paragraph [0030] continues:
"The PreF antigens are favorably used for the prevention and/or treatment of RSV infection. Thus, another aspect of this disclosure concerns a method for eliciting an immune response against RSV. The method involves administering an immunologically effective amount of a composition containing a PreF antigen to a subject (such as a human or animal subject). Administration of an immunologically effective amount of the composition elicits an immune response specific for epitopes present on the PreF antigen. Such an immune response can include B cell responses (e.g., the production of neutralizing antibodies) and/or T cell responses (e.g., the production of cytokines). Favorably, the immune response elicited by the PreF antigen includes elements that are specific for at least one conformational epitope present on the prefusion conformation of the RSV F protein. The PreF antigens and compositions can be administered to a subject without enhancing viral disease following contact with RSV. Favorably, the PreF antigens disclosed herein and suitably formulated immunogenic compositions elicit a Th1 biased immune response that reduces or prevents infection with a RSV and/or reduces or prevents a pathological response following infection with a RSV."
Terms
- EP258 then proceeds to define certain terms. Two of the definitions are particularly material, which I quote below, but I do not lose sight of the fact that a number of terms used (or alluded to) in the claims are specifically defined, including 'recombinant' in [0046], 'heterologous' in [0047], 'antigen' in [0050], 'immunogenic composition' in [0052] and 'pharmaceutically acceptable' in [0056] and 'prevents' in [0058]. Whilst it may be said that some of these definitions are unnecessary (because the defined meaning is also the accepted technical meaning), this set of definitions illustrates very clearly that the patentee is using these terms in a precise way.
[0039] The term "F protein" or "Fusion protein" or "F protein polypeptide" or Fusion protein polypeptide" refers to a polypeptide or protein having all or part of an amino acid sequence of an RSV Fusion protein polypeptide."
[0043] The term "polypeptide" refers to a polymer in which the monomers are amino acid residues which are joined together through amide bonds. The terms "polypeptide" or "protein" as used herein are intended to encompass any amino acid sequence and include modified sequences such as glycoproteins. The term "polypeptide" is specifically intended to cover naturally occurring proteins, as well as those which are recombinantly or synthetically produced. The term "fragment," in reference to a polypeptide, refers to a portion (that is, a subsequence) of a polypeptide. The term "immunogenic fragment" refers to all fragments of a polypeptide that retain at least one predominant immunogenic epitope of the full-length reference protein or polypeptide. Orientation within a polypeptide is generally recited in an N-terminal to C-terminal direction, defined by the orientation of the amino and carboxy moieties of individual amino acids. Polypeptides are translated from the N or amino-terminus towards the C or carboxy-terminus."
PreF antigens
- PreF antigens are then addressed. Paragraph [0060] essentially explains that in nature the RSV protein is expressed in the F0 polypeptide, so it is expressed as a single polypeptide, and then it explains how it is cleaved and how the conformation changes:
"In nature, the RSV F protein is expressed as a single polypeptide precursor 574 amino acids in length, designated F0. In vivo, F0 oligomerizes in the endoplasmic reticulum and is proteolytically processed by a furin protease at two conserved furin consensus sequences (furin cleavage sites), RARR109 (SEQ ID NO:15) and RKRR136 (SEQ ID NO:16) to generate an oligomer consisting of two disulfide-linked fragments. The smaller of these fragments is termed F2 and originates from the N-terminal portion of the F0 precursor. It will be recognized by those of skill in the art that the abbreviations F0, F1 and F2 are commonly designated F0, F1 and F2 in the scientific literature. The larger, C-terminal F1 fragment anchors the F protein in the membrane via a sequence of hydrophobic amino acids, which are adjacent to a 24 amino acid cytoplasmic tail. Three F2-F1 dimers associate to form a mature F protein, which adopts a metastable prefusogenic ("prefusion") conformation that is triggered to undergo a conformational change upon contact with a target cell membrane. This conformational change exposes a hydrophobic sequence, known as the fusion peptide, which associates with the host cell membrane and promotes fusion of the membrane of the virus, or an infected cell, with the target cell membrane.".
- Paragraph [0061] defines the F1 fragment:
"The F1 fragment contains at least two heptad repeat domains, designated HRA and HRB, and situated in proximity to the fusion peptide and transmembrane anchor domains, respectively. In the prefusion conformation, the F2-F1 dimer forms a globular head and stalk structure, in which the HRA domains are in a segmented (extended) conformation in the globular head. In contrast, the HRB domains form a three-stranded coiled coil stalk extending from the head region. During transition from the prefusion to the postfusion conformations, the HRA domains collapse and are brought into proximity to the HRB domains to form an anti-parallel six helix bundle. In the postfusion state the fusion peptide and transmembrane domains are juxtaposed to facilitate membrane fusion."
- [0062] is relevant to the insufficiency case.
"Although the conformational description provided above is based on molecular modelling of crystallographic data, the structural distinctions between the prefusion and postfusion conformations can be monitored without resort to crystallography. For example, electron micrography can be used to distinguish between the prefusion and postfusion (alternatively designated prefusogenic and fusogenic) conformations, as demonstrated by Calder et al., Virology, 271:122-131 (2000) and Morton et al., Virology, 311:275-288. The prefusion conformation can also be distinguished from the fusogenic (postfusion) conformation by liposome association assays as described by Connolly et al., Proc. Natl. Acad. Sci. USA, 103:17903-17908 (2006). Additionally, prefusion and fusogenic conformations can be distinguished using antibodies (e.g., monoclonal antibodies) that specifically recognize conformation epitopes present on one or the other of the prefusion or fusogenic form of the RSV F protein, but not on the other form."
- [0063] is relevant to stability, explaining also the purpose of stabilisation:
"The PreF antigens disclosed herein are designed to stabilize and maintain the prefusion conformation of the RSV F protein, such that in a population of expressed protein, a substantial portion of the population of expressed protein is in the prefusogenic (prefusion) conformation (e.g., as predicted by structural and/or thermodynamic modeling or as assessed by one or more of the methods disclosed above). Stabilizing modifications are introduced into a native (or synthetic) F protein, such as the exemplary F protein of SEQ ID NO:2, such that the major immunogenic epitopes of the prefusion conformation of the F protein are maintained following introduction of the PreF antigen into a cellular or extracellular environment (for example, in vivo, e.g., following administration to a subject)."
- [0064]-[0068] then describe various stabilising modifications. First (in [0064]) a heterologous trimerization domain which is predicted to compensate for the HRB instability, helping to stabilize in the prefusion conformation. [0065] explains that, in order to stabilize HRB even more, the leucine residue at position 512 can be substituted with lysine to improve the coiled coil hydrophobic residue periodicity. Second, in [0066] it is explained that pep27 can be removed because it creates a large unconstrained loop between F1 and F2 which does not contribute to stabilisation of the prefusion state. [0067] describes the third suggested method and it concerns the deletion of the furin cleavage sites. This paragraph is important in identifying the purpose of that deletion, but also the importance of preventing membrane access to the fusion peptide:
[0067] Third, one or both furin cleavage motifs can be deleted. With this design, the fusion peptide is not cleaved from F2, preventing release from the globular head of the prefusion conformer and accessibility to nearby membranes. Interaction between the fusion peptide and the membrane interface is predicted to be a major issue in the prefusion state instability. During the fusion process, interaction between the fusion peptide and the target membrane results in the exposure of the fusion peptide from within the globular head structure, enhancing instability of the prefusion state and folding into post-fusion conformer. This conformation change enables the process of membrane fusion. Removal of one or both of the furin cleavage sites is predicted to prevent membrane accessibility to the N-terminal part of the fusion peptide, stabilizing the prefusion state.
Adjuvants
- There is then a long discussion of immunogenic compositions and methods at [0106] to [0148] including pharmaceutically acceptable carriers and excipients.
- [0149] is a long paragraph containing what appear to be the original consistory clauses for the claims. Claim 1 as granted is a combination of claims 1, 4 and 5 as set out in [0149]. I have to return to [0149] later.
- The description of the 8 Examples starts at [0151]. Examples 1 to 3 relate to exemplary PreF antigens and production, purification and characterization of the PreF recombinant protein. Examples 4 to 8 relate to various immunogenic assays.
Example 1
- Example 1, at [0151] and [0152] describes the modifications that were made to the particular "PreF" antigens tested in the experiments modified as compared to a native RSV F protein in order to stabilise the protein.
- Then [0152]: "FIG. 1A and B", "schematically illustrate features of the RSV F0 and exemplary PreF recombinant antigens. FIG. 1A is a representation of the RSV F0 protein", and it goes through and discusses that all the way down to line 45. It discloses that the F0 protein is proteolytically processed and glycosylated.
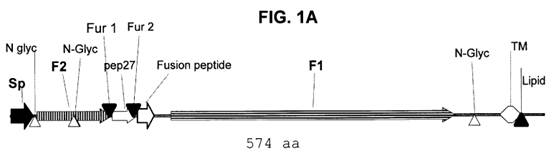
- Fig 1B is a schematic illustration of two exemplary RSV Prefusion F (PreF) antigens. [0152] explains:
"To construct the PreF antigen, the F0 polypeptide was modified to stabilize the prefusion conformation of the F protein, thereby retaining the predominant immunogenic epitopes of the F protein as presented by the RSV virus prior to binding to and fusion with host cells. The following stabilizing mutations were introduced into the PreF antigen relative to the F0 polypeptide. First, a stabilizing coiled-coil domain was placed at the C-terminal end of the extracellular domain of the F0 polypeptide, replacing the membrane anchoring domain of F0. Second, the pep27 peptide (situated between the F2 and F1 domains in the native protein) was removed. Third, both furin motifs were eliminated. In alternative embodiments (designated PreF _V1 and PreF_V2), an immunologically active portion (e.g., amino acids 149-229) of the RSV G protein was added to the C-terminal domain.
- These primary and alternative embodiments are illustrated in Fig.1B:
- Although this is not explained in the context of either Example 1 or Fig 1B, the Skilled Structural Biologist would note that, following removal of pep27, the remaining ends must be joined to form the single polypeptide shown in either of the embodiments in Fig 1B. Professor Weissenhorn pointed out that an engineered lysine residue (K) has been inserted to connect the F2 and F1 regions (see further below).
Example 2
- Example 2 describes the production and purification of the antigen. Nothing turns on it.
Example 3
- Example 3 characterises the PreF recombinant protein produced. The way in which this was characterised was to run it through, at [0157], "... by asymmetrical field flow fractionation (AFF-MALS) and compared to a chimeric antigen including RSV F and G protein components." The AFF-MALS technique resolves molecules in a mixture according to their hydrodynamic size. The construct of Example 1 (PreF) is compared with the FG control.
- Figs 2A and 2B are the figures that show the results of this comparison. AFF-MALS can distinguish between whether the FG or the prefusion respectively are in the aggregated form or whether they are in the trimerised form. At [0158] it is noted that "FIG. 2B shows that the purified PreF protein is folded in his trimeric form to a proportion of 73% in PBS buffer. 10% of the material is found as 1000 to 20 000 KDa oligomers. These results indicate that the recombinant PreF protein expressed in CHO cells is folded as a trimer as predicted for the native state."
- The FG control comprises the extracellular portions of the F and G proteins. The F antigen in the FG construct has its furin cleavage sites intact, so will be cleaved and therefore has the capacity to assume the postfusion conformation. This results in the fusion peptide becoming exposed which leads to aggregation. This can be seen in the data in the top trace in Figure 2 ("FG") where 95% are "high molecular weight aggregates". If the construct is in prefusion form then the fusion protein is not exposed and the antigen is not prone to aggregation.
Example 4
- Example 4 describes in vitro neutralization inhibition by the PreF antigen. In summary this is a survey of the type of antibodies which are found in a human population. The data indicates that in a greater proportion of patients there are more neutralising antibodies to the PreF form of the F antigen (solid black histogram) than to FG. The Patents state that Exemplary results shown indicate that PreF is superior to FG in 16/21 donors.
- Example 4 compares the ability of PreF and a control FG chimeric protein to absorb neutralising antibodies from human serum samples.
Example 5
- Example 5 describes an experiment which demonstrates that the PreF antigen is immunogenic. Mice were immunised with the PreF construct with and without a Th1 adjuvant. Adjuvants were typically used to enhance the immunogenicity of a vaccine. The data are at Figure 4 and show that mice produced antibodies to both strains of RSV, being RSVA and RSVB.
Example 6
- Example 6 demonstrates that the PreF antigen elicits neutralising antibodies. Infectious doses of RSV were added to serum samples from mice immunized as described in Example 5, and the mixture incubated for 5 to 6 days. The results in Figures 5A and 5B show neutralising antibody titres. The neutralising titre of a serum sample is usually calculated as the reciprocal of the dilution of the serum that neutralises 50% or 60% of the virus.
Example 7
- This describes an experiment which demonstrated the ability of PreF to protect mice from RSV infection. Example 7 introduces an alternative adjuvant to those used in the previous Examples which indicates that different mice were used. Good protection by PreF adjuvanted with each adjuvant against RSV challenge was shown. The adjuvant control with no PreF antigen resulted in the same degree of protection as a dose of the PreF antigen without an adjuvant (Fig 6B).
Example 8
- Example 8 investigates whether PreF causes eosinophil recruitment, which was considered a marker for enhanced lung pathology as had been observed from the unsuccessful 1960s clinical trial. Mice were immunized with PreF, glutaraldehyde-treated PreF (which deliberately aggregates the protein) and FG Rix, each without adjuvant, and challenged with RSV three weeks later. Levels of eosinophils from collected lung cells are recorded.
- Figure 7 reveals that, whereas both glutaraldehyde-treated PreF and FG Rix induced eosinophils, the PreF antigen did not.
- Both experts criticised the lack of a positive control despite the Example using both FG Rix (containing an unmodified F protein) and a deliberately aggregated PreF antigen.
Sequence information
- The specification concludes with many pages of sequence listings. Professor Weissenhorn drew attention in particular to SEQ ID NO:2 Amino acid sequence of RSV reference F protein precursor F0 and SEQ ID NO:6 Amino acid sequence of PreF analog and I also note SEQ ID NO:15 Furin cleavage site RARR and SEQ ID NO:16 Furin cleavage site RKRR.
- Professor Weissenhorn undertook a comparison between SEQ ID NO:2 and SEQ ID NO:6 to find out exactly which modifications were made for the PreF analog. His comparison revealed that although three modifications are described in the context of Example 1, in fact five modifications have been made. The additional two modifications are described by Professor Weissenhorn as follows:
'(i) that the PreF sequence in SEQ ID NO:6 includes an engineered lysine residue which connects the F2 and F1 regions. It is at position 106 of the SEQ ID NO: 6 sequence. Given the pep27 region is removed and furin cleavage sites have been deleted, the lysine residue was engineered into the sequence to link the F2 and F1 sequences together. That same modification is described by the patent as "another example of a stabilizing mutation" at paragraph [0018]. Because the lysine residue might be exposed, it could contribute to solubility of the protein, being a positively charged amino acid, and it may also serve as a "short" linker that contributes to stability because its presence may prevent distorting the region linking F2 and F1.
(ii) The amino acid substitution at position 482, referred to in paragraph [0065] as another stabilizing mutation, is also present in the SEQ ID NO:6 sequence. A modification to improve periodicity in the HRB region could also potentially have an impact on the stability of the protein.'
- Accordingly, if the SEQ NO:6 represents the sequence of the PreF antigens tested in the Examples, they comprise five stabilising modifications. However, Professor Weissenhorn pointed out that (a) the Skilled Structural Biologist would not know whether the modifications had been incorporated into the PreF antigens tested in the Examples and (b) it would not be possible to assess the relative contributions of the trimerization domain on its own, absent the other modifications, without proper controls.
- For EP258: claims 1, 5 and 8 as amended (or claims 1, 5 and 9 as unamended) are said to be independently valid. The terms in dispute are in italics:
1. A recombinant respiratory syncytial virus (RSV) antigen which assembles into a trimer, comprising a soluble F protein polypeptide comprising an F2 domain and an F1 domain of an RSV F protein polypeptide and comprising an amino acid sequence comprising a heterologous trimerization domain positioned C-terminal to the F1 domain that stabilizes the prefusion conformation of the F protein.
5. An immunogenic composition comprising the recombinant RSV antigen of any one of claims 1-3, and a pharmaceutically acceptable carrier or excipient.
9. The recombinant RSV antigen of any one of claims 1-3 or the immunogenic composition of any one of claims 5-7 for use in the prevention or treatment of RSV-associated diseases.
- For EP 710: claims 1, 10, 22, 23 and 24 are said to be independently valid:
1. A recombinant respiratory syncytial virus (RSV) antigen comprising a soluble F protein polypeptide comprising an F2 domain and an F1 domain of an RSV F protein polypeptide, wherein there is no furin cleavage site between the F2 domain and the F1 domain, and wherein the polypeptide further comprises a heterologous trimerization domain positioned C-terminal to the F1 domain.
10. An immunogenic composition comprising the recombinant RSV antigen of any one of claims 1-9, and a pharmaceutically acceptable carrier or excipient.
22. The recombinant RSV antigen of any one of claims 1-9 or the immunogenic composition of any one of claims 10-15 for the prevention or treatment of RSV-associated diseases.
23. The recombinant RSV antigen of any one of claims 1-9, wherein the recombinant RSV antigen is stabilized in the prefusion conformation of the F protein.
24. The recombinant RSV antigen of any one of claims 1-9 or 23, wherein the recombinant RSV antigen is a soluble RSV F protein polypeptide stabilized in the prefusion conformation of the F protein.
- The claims alleged to be infringed are:
i) For EP 258: claims 1, 5, 7 and 8 as amended (or claims 1, 5, 8 and 9 as unamended);
ii) For EP 710: claims 1-4, 10, 11, 14, and 19-24.
- My task is to undertake a 'normal construction' of the claims. It is unnecessary for me to set out the standard authorities but I have in mind Actavis v Lilly [2017] UKSC 48, Icescape v Ice-World [2019] FSR 5 and Liqwd Inc v L'Oreal UK Ltd [2018] EWHC 1394 (Pat), Birss J.
- As mentioned above, the parties are in dispute over the meaning of the terms "stabilizes" and "polypeptide". The meaning of "stabilizes" is relevant to validity, that of "polypeptide" to infringement. I can decide these points here, because the CGK disputes which remain to be concluded do not affect them.
- Before addressing those specific issues, there are some general features of both sets of claims which should be noted. Whereas the Examples concern one particular PreF construct with at least 3 but possibly 5 modifications, the claims are limited to one or two of the 3 modifications.
- GSK contend this term should be interpreted as follows:
"a greater proportion of the F protein is in the prefusion conformation than would be the case in a preparation of an antigen in which the F protein does not have a heterologous trimerization domain positioned C-terminal to the F1 domain."
- GSK suggest that the Patents advance various methods of achieving stabilisation one of which is using a heterologous trimerization domain. To fall within the scope of the claim the antigen must comprise a heterologous trimerization domain positioned C-terminal to the F1 domain. This will 'stabilise' the antigen in the prefusion conformation. This does not need to be the only modification made to achieve stabilisation: in practice the skilled person seeking to put the invention into effect may additionally use the other modifications which are proposed by the Patents.
- Pfizer has relied on GSK's construction and approached its evidence on the same basis, but notes that "stabilizes" can be context dependent, relying on the following points:
i) In an immunological context, it would be understood that stabilizing the prefusion conformation would require that the RSV F protein be in the native (prefusion) conformation long enough to initiate an immune response specific for the epitopes on the prefusion conformation of RSV in a vaccinated individual.
ii) In a structural/molecular biology context, it would be understood that any definition of the term depends on the context in which it was used. Although there was no established definition in the field at the time, it was often used in relative terms to compare protein A and its modified version thereof with respect to their stability.
iii) It is important to assess issues of construction in the appropriate context. At its broadest, this means in the context of the patent as a whole, when read through the eyes of the skilled person armed with the common general knowledge.
- I agree that "stabilizes" is deliberately broad and not a term of art given its use in a number of different contexts, both in the specification and in various claims. Furthermore, the Skilled Team would be likely to conclude that if the trimerization domain is the only stabilizing modification, the degree of stability conferred would be modest (possibly extremely modest).
- Although the Court is obliged to construe the Patent(s) as if the alleged infringer had never existed, as in many cases, the issue is best explained by reference to the alleged infringement.
- All the claims require an RSV antigen 'comprising a soluble F protein polypeptide comprising an F2 domain and an F1 domain of an RSV F protein polypeptide'.
-
 An image of the primary sequence of Pfizer's RSVpreF product is as follows, in which the black lines represent covalent disulphide bonds.
An image of the primary sequence of Pfizer's RSVpreF product is as follows, in which the black lines represent covalent disulphide bonds.
- There is no dispute that:
i) Pfizer's RSVpreF product comprises an F2 domain and F1 domain.
ii) As occurs in the native F protein, pep27 section of the F0 polypeptide has been removed by cleavage at the two furin cleavage sites.
iii) Also as in the native F protein, the F1 and F2 domains remain linked by covalent disulphide bonds.
iv) However, as GSK accepted, a non-native disulphide bond has been added - it is the third bond from the left.
v) The RSVPreF product does not comprise a single stream of amino acids joined together through amide bonds.
- Thus, the issue is whether the term 'polypeptide' requires a single chain of amino acids joined together through amide bonds or whether it should be interpreted more broadly so that it also includes dimers made up of more than one polypeptide joined together other than by such amide bonds.
Pfizer's arguments
- Pfizer's argument is simple. The term 'polypeptide' is defined in the Patents at [0043] in the following way: 'The term "polypeptide" refers to a polymer in which the monomers are amino acid residues which are joined together through amide bonds.'
- Pfizer also point out that this is also its standard technical meaning and furthermore that the Patents appear to be scrupulous in using the terms 'polypeptide' and 'domain' entirely in accordance with their accepted technical meanings. For what it is worth, their position was supported by Professor Weissenhorn in his evidence, where he took the view that the Pfizer RSVpreF comprises two polypeptides.
- Pfizer also submitted that a single polypeptide was an important feature of the invention. As I understood the logic behind this submission, it was that the removal of the pep27 section would ordinarily result in the separation of the F0 precursor into two polypeptides, F1 and F2. Yet the Patentee has engineered their joinder back into a single polypeptide, by the addition of a lysine residue at position 106 of SEQ ID:6.
GSK's arguments
- In their closing, GSK devoted a number of paragraphs to this issue, but, conspicuous by its absence was any reference to the definition in the Patents at [0043].
- In essence, GSK's arguments fell into the following three categories:
i) What they contended was the 'ordinary English sense' of the term.
ii) The combinations of modifications described in the Patents.
iii) Technical purpose.
- On the first point, GSK relied on the view of Professor Wilkinson that Professor Weissenhorn's view was 'unusually pedantic'. He said that common usage of "polypeptide" is interchangeable with subunit, chain and protein, and so can encompass sequences of protein which are bonded by disulphide bonds.
- GSK accepted that 'polypeptide' encompasses a construct which is a single string of amino acids, but nonetheless contended the term is a broad one which, they said, naturally gets used to describe all these sorts of constructs.
- GSK's starting point was the definitions in [0039] (set out above). Their contention was that [0039] was neutral as to this issue on construction but that it showed the terms are used as a matter of ordinary English. GSK also relied on various examples of how authors have used the term 'polypeptide' in certain papers, on the basis of which they contend that the 'ordinary English sense' therefore supported GSK's case.
- In cross-examination, two documents were put to Professor Weissenhorn to show the authors of each document using 'polypeptide' in a wider sense.
- First, Buckland (1987) [DXX-WW/1] was a primary research paper concerning the fusion protein in measles virus, another paramyxovirus and with a fusion protein which undergoes furin cleavage and a conformational change akin to RSV F. The authors state (sentences bridging pages 1-2):
"The F protein is responsible for cell penetration ... It is synthesized as a precursor polypeptide F0 and is cleaved by cellular proteases to give the biologically active polypeptide F1,2." (GSK's emphasis added)
- GSK's contention was that the authors were describing a precisely analogous construct (namely, a cleaved F protein comprising an F1 and F2 chain held together by disulphide bonds) as a singular "polypeptide". Professor Weissenhorn accepted this.
- Second, a textbook from 2020 entitled "Frontiers in Protein Structure, Function, and Dynamics" [DXX-WW/2] stated in relation to the SDS-PAGE characterisation technique:
"Molecular masses of the protein subunits can also be determined by SDS-PAGE as SDS disrupts the non-covalent interactions among polypeptides. The link between subunits in a polypeptide formed by disulfide bonds can be determined using SDS-PAGE via preparing protein samples both in the absence and presence of reducing agent 2-mercaptoethanol which breaks the disulfide bond." (GSK's emphasis added)
- GSK contended that although Professor Weissenhorn initially quibbled with this passage, he accepted on reflection that this passage envisages being able to detect the molecular mass of the protein subunits - i.e., that the single polypeptide is formed of multiple chains which are linked by scissile disulphide bonds.
- In my judgment, these examples carry no weight whatsoever. It is not surprising that diligent searching can reveal some uses of 'polypeptide' in a broad sense, in different contexts. The point is that neither set of authors included in their document the definition which the Patents contain in [0043].
- The second strand of GSK's argument depends on the combinations of modifications which are explained in the Patents. Thus, as GSK submitted, the Patents make clear that their proposed stabilising modifications can be implemented individually or in combination (see [0015] line 8). These are described in detail in paragraphs [0064]-[0067] and include the addition of a heterologous stabilizing domain (at [0064]) and/or the deletion of "one or both" furin cleavage sites (at [0067]).
- In cross-examination, Professor Weissenhorn was shown some diagrams which set out depictions of the constructs which are produced by selecting combinations of these modifications. He agreed that, if only a trimerization domain is added but the furin cleavage sites are left in place, the resulting mature construct will be as shown in [DXX-WW/4/1]. It shows two chains linked by disulphide bonds.
- GSK also relied on [0014] of EP258 (set out above) contending that it envisages making one or more of the modifications the Patents later describe, and therefore contemplates a construct of the sort in [DXX-WW/4/1] - i.e. a construct of two chains linked by disulphide bonds.
- GSK even went as far as to submit that:
'In relation to that construct, the Patents use the term "soluble F protein polypeptide" which is the same feature as used in the claims. Therefore, the patentee is itself using the term "polypeptide" to encompass constructs such as Pfizer's RSVpreF product.'
- GSK made the same point based on [0149] which describes further aspects of the invention. The first and second sub-paragraphs to [0149] state (with GSK's emphasis added):
"1. A recombinant respiratory syncytial virus (RSV) antigen comprising a soluble F protein polypeptide comprising at least one modification that stabilizes the prefusion conformation of the F protein.
2. A recombinant respiratory syncytial virus (RSV) antigen comprising a soluble F protein polypeptide, which F protein polypeptide comprises at least one modification selected from:
(i) an addition of an amino acid sequence comprising a heterologous trimerization domain;
(ii) a deletion of at least one furin cleavage site;
(iii) a deletion of at least one non-furin cleavage site;
(iv) a deletion of one or more amino acids of the pep27 domain; and
(v) at least one substitution or addition of a hydrophilic amino acid in a hydrophobic domain of the F protein extracellular domain."
- GSK submitted that these paragraphs put beyond doubt that the patentee uses "soluble F protein polypeptide" to encompass constructs which, when expressed, will be composed of an F1 chain and an F2 chain linked by disulphide bonds and not continuously bonded through only peptide bonds.
- All these arguments confuse what the Patents disclose with what they actually claim. It is plain that far more is disclosed than is claimed in either EP258 or EP710 - indeed that is what lies behind Pfizer's claim for Arrow relief.
- It is true that there appears to be a conflict between the definition of 'polypeptide' in [0043] and certain of the combinations of modifications disclosed in other paragraphs. What is clear, however, is that the claims are much more limited than the disclosure, being limited (in the case of EP258) to stabilisation by the addition of a heterologous trimerization domain and (in the case of EP710), to that plus the absence of furin cleavage sites.
- GSK's third point was based on technical purpose. GSK submitted there was no reason why the distinction between the two constructions of 'polypeptide' is one that matters at the level of the invention, with GSK contending that the purpose of the invention was to raise an immune response by presenting an F protein stabilised in the prefusion state. However, this purpose is at a level well above any of the claims.
- At the correct level - that of the claims - in my judgment the conclusion is inescapable that the Patentee was using 'polypeptide' in accordance with the internal dictionary meaning set out in [0043].
- On this issue it is plain that each Patent contains its own dictionary, providing a precise and technically accurate definition of the term 'polypeptide' for all purposes (i.e. both infringement and validity). It is clear that the Patent uses that and other terms accurately (e.g. the F1 and F2 domains). The Patentee chose to include that definition. It could have defined the term more broadly, but that might have caused other issues and it chose not to do so.
- A purposive interpretation of the term reinforces the importance of the definition given expressly by the Patents themselves. Ignoring that meaning by extending it to include antigens that are not single polypeptides goes against the central teaching relevant to the claims. I therefore interpret "polypeptide" to refer to "a polymer in which the monomers are amino acid residues which are joined together through amide bonds".
- The basic legal principles were not in dispute between the parties. As is well-known, the Supreme Court decision Actavis v Eli Lilly [2017] UKSC 48 sets out how to assess infringement by equivalents. Having reviewed the law on infringement and applying Actavis, the Court of Appeal in Icescape Limited v Ice-World International BV & Ors [2017] EWHC 42 (Pat) summarised in [66] the main steps in the approach for assessing infringement, as follows:
"66. The whole approach to interpretation and scope of protection therefore involves the following steps, considered through the eyes of the notional addressee:
(i) Does the variant infringe any of the claims as a matter of normal interpretation?
(ii) If not, does the variant nevertheless infringe because it varies from the invention in a way or ways which is or are immaterial? This is to be determined by asking these three questions:
(a) Notwithstanding that it is not within the literal (that is to say, I interpolate, normal) meaning of the relevant claim(s) of the patent, does the variant achieve substantially the same result in substantially the same way as the invention, i.e. the inventive concept revealed by the patent?
(b) Would it be obvious to the person skilled in the art, reading the patent at the priority date, but knowing that the variant achieves substantially the same result as the invention, that it does so in substantially the same way as the invention?
(c) Would such a reader of the patent have concluded that the patentee nonetheless intended that strict compliance with the literal meaning of the relevant claim(s) of the patent was an essential requirement of the invention?"
- As GSK submitted, the Actavis questions require a proper characterisation of the inventive concept.
- GSK relied in particular on the analysis of Meade J. in Optis Cellular Technology LLC v Apple Retail UK Limited [2021] EWHC 1739 (Pat) (Trial B), where he addressed the Actavis questions in the context of an allegation of anticipation by equivalence. As GSK submitted, Meade J. had to resolve a dispute between the parties as to the right level of generality at which to consider the first Actavis question. The parties' rival characterisations were set out in paragraphs 257 and 258 of the judgment:
"257. Apple relies on the "result" being tracking the same sets of resources, namely sequence number usage and buffer memory usage. Its submissions essentially treated this as the "way" as well. Apple's approach was based on, and arose from, evidence Mr Kubota had given on equivalence for the purpose of infringement/essentiality, where he referred to tracking those resources.
258. Optis disagreed. It pointed out that in addition to tracking those resources, Mr Kubota had referred to simplicity of implementation and (claim 9) avoidance of superfluous polling. It pointed out that InterDigital is more complex (as windows based mechanisms will always be) and does not avoid superfluous polling. I agree with those points, and I have already analysed why above."
- The judge resolved this difference by having regard to the "results" that were identified in the specification:
"260. In the present case, I have no hesitation in reaching the conclusion that the right level of generality, and the result to be considered, is as Optis says, and Apple's argument is at the wrong level of generality. Simplicity and avoiding superfluous polling are both relevant "results" and they are achieved by the specific use of counting (as I have construed it) and immediate resets, while (although this is not necessary to my conclusion) omitting the use of status reports. It is justifiable to regard these as the "result" and as being at the right level of generality because they are flagged in the specification. So Apple fails on question 1, for all the disputed integers." (my emphasis).
- GSK submitted the characterisation of the "result" in the specification is relevant.
- I have underlined what appears to be the key sentence in [260], but I do not consider that Optis establishes any principle to the effect that any 'result' flagged in the specification is at the right level of generality. From my reading of Optis, the teaching in the specification appears to have been specific to what was claimed.
- Key to claim 1 was 'counting the number of transmitted data units'. [0017] in particular explained how the claimed method avoided superfluous polling and made for a simpler system. As the Judge said:
'110. Paragraphs [0009] and [0010] of the Patent discuss counter-based and windows-based mechanisms in such a way as to contrast them.'
- One way to explain the contrast was as follows:
'116. Another way of looking at this is that counter-based mechanisms operate in units of PDUs and bytes, whereas windows-based mechanisms operate in terms of percentages, because they have regard to how many PDUs or bytes there are in the buffer compared with the maximum PDUs or bytes allowed.
117. In my view, "counting" in claim 1 is clearly intended to take its meaning from this rather particular context. It means maintaining a count of transmitted PDUs and bytes, in such units.'
- Resetting was the subject of claims 6 and 9. Meade J. quoted [0046] at [136], saying at [137] it had an obvious echo of [0017], and explained in [138] that resetting also prevented superfluous polling.
- The alleged anticipation was Interdigital which was a Tdoc describing a window-based polling mechanism. There was no anticipation because the mechanism did not involve 'counting' as the Judge had construed that term.
- Against that backdrop, it can be seen that Apple's submission that the 'result' was 'tracking the same sets of resources, namely sequence number usage and buffer memory usage' lay at a level of generality far higher than the claims. It appears to me that the judge accepted Optis' argument because the simplicity of operation and avoidance of superfluous polling were results directly linked to the claimed features.
- This diversion into Optis v Apple merely confirms that the only principle is that, when assessing the result and the way for Actavis question 1, one has to have regard to the correct level of generality and that is at the level of the particular claim under consideration.
- GSK alleges that Pfizer's product, RSVpreF, infringes each of EP 258 and EP 710. RSVpreF contains RSV F antigens which the PPD accepts are in the prefusion form.
- I have set out the features of the RSVPreF protomer above. I should mention that the RSVPreF is a bivalent vaccine product comprising two variants RSVPreF-A and RSVPreF-B, but I am told there is no material difference between them for present purposes.
- I remind myself that the claims alleged to be infringed are:
i) EP 258: claims 1, 5, 7 and 8 as amended (or claims 1, 5, 8 and 9 as unamended);
ii) EP 710: claims 1-4, 10, 11, 14, and 19-24.
- All the claims of EP258 require a trimerization domain positioned C-terminal to the F1 domain that stabilizes the prefusion conformation of the F protein, whereas that combination of the trimerization domain which stabilises the prefusion conformation only comes in claims 23 and 24 of EP710. The earlier claims of EP710 claim either an antigen or an immunogenic composition by way of structural features.
- The key issue on normal infringement concerns this expression "a soluble F protein polypeptide comprising an F2 domain and an F1 domain" and arises on both EP 258 and EP 710.
EP258
- Pfizer's RSVpreF product comprises an F2 domain and an F1 domain. As occurs in the native F protein, the pep27 section of the F0 polypeptide has been removed by cleavage at the two furin cleavage sites. However, and also as in the native F protein, F1 and F2 remain linked by covalent disulphide bonds.
- Based on my construction of 'polypeptide' as set out above, Pfizer's RSVpreF does not infringe EP258. It does not comprise "a soluble F protein polypeptide comprising an F2 domain and an F1 domain" but instead "two polypeptides".
- Pfizer's further point is that their RSVPreF does not have an adjuvant, but that relates to insufficiency.
EP710
- The "polypeptide" point applies equally to EP710, but the inappropriate nature of GSK's construction is made even more clear in light of the further requirement that there be no furin cleavage site "between" F1 and F2. The absence of a cleavage site "between" the domains makes no sense unless the F1 and F2 domains are part of a single polypeptide. In addition, it also highlights the purpose of the absence, namely to avoid cleavage of that polypeptide.
- Before addressing the specific Actavis questions, it is worth identifying the key issue on both Patents: the appropriate level of generality at which to identify the relevant inventive concept and hence the relevant 'result' and 'way'. As one might expect from GSK's reliance on Optis, they seek to rely on effects described in the specification.
EP 258
- On Question 1 there was a degree of common ground namely: 1) the stabilisation of the prefusion form is of central importance to the inventive concept and 2) if PreF is stabilised then the access of the fusion peptide to the membrane is restricted.
- Pfizer's position is that the claims of EP258 (and indeed EP710) claim particular ways in which such restriction of access is to be achieved, namely the use of single polypeptide and the addition of the further specific stabilising features claimed. Pfizer therefore suggests the inventive concept is the restriction of access by the fusion peptide to the membrane by the mechanisms claimed.
- In light of the above, Pfizer submits that the answer to Actavis Question 1 is no, as the way in which the fusion peptide is prevented from accessing the membrane (in RSVPreF) and the way in which the product of the patent works would not be thought to be achieving that end in the same way. In terms of chemistry, they would be seen to be done differently, in particular:
i) The RSVPreF product achieves stabilisation not through reducing the chance of the change in conformation in a single polypeptide, but through preventing the change in conformation via the use of the non-native disulphide bond together with the other features such as the C terminal domain and the two other mutations.
ii) The claimed invention of EP258 achieves stabilisation of a single polypeptide by a trimerization domain. RSVPreF achieves stabilisation by cleaving the polypeptide and forming a dimer of which the two parts are held together by a non-native disulphide bond and other modifications including the trimerization domain.
- Professor Wilkinson opined that the RSVPreF 'achieves substantially the same result in substantially the same way as the invention' based on his view that both removal of the furin cleavage sites and the addition of a non-native disulphide bond constrain the fusion peptide. Professor Weissenhorn disagreed and gave detailed reasons why. Whilst he accepted that both approaches may stabilize the prefusion conformation and involve the fusion peptide, he was of the view that they do so in very different ways.
- For EP258, he referred to the statement in [0067] to the effect that in the prefusion conformation the fusion peptide is buried within the globular head structure and that during the fusion process the fusion peptide becomes exposed, allowing interaction with the target membrane. He said the skilled structural biologist would know that as part of this conformational rearrangement the fusion peptide needs to change its location so that it can access the target membrane. He stated that the modification to the native F protein described in the Patents works by constraining the fusion peptide within a single, continuous polypeptide chain in which the F2 and F1 domains are linked by a peptide bond and further stabilizing the protein by adding a C-terminal trimerization domain to the F1 domain. He was of the view that, by keeping the fusion peptide within a single polypeptide chain, the fusion peptide is not so free to move and the activation energy required for the protein to refold into the conformation increases.
- He accepted that this modification may contribute to the stabilisation of the prefusion conformation of the F protein because membrane accessibility to the N-terminal part of the fusion peptide is hampered. However, his point was that reducing membrane accessibility in this way does not hold the prefusion conformation in position or prevent the postfusion conformation forming, just that it makes the prefusion conformation more stable.
- Turning to the RSVPreF, Professor Weissenhorn said the construct works in a different way, relying on four modifications but placing by far the greatest emphasis on the first, being the introduction of an additional non-native disulphide bond between positions C103 (F2) and C148 (part of the fusion peptide within the F1 domain). He said this bond locks the two polypeptide chains together in the prefusion conformation and prevents a transition to the postfusion form. A switch to the postfusion form would require the breaking of this disulphide bond. In consequence, he was of the view that the skilled structural biologist would understand the RSVPreF product to be stabilised in a materially different way.
- Professor Weissenhorn also drew attention to three further modifications: 1) a cavity filling change (Ser190 to IIe), the removal of a negative charge (Asp486 to Ser) and a C-terminal fusion of the trimeric foldon domain to the F1 polypeptide.
- The Professor was asked to prepare a schematic diagram to show the features of the RSVPreF which contributed to stabilisation, together with an indication of the position of the fusion peptide buried inside the globular structure of the F1 and F2 polypeptides in the prefusion conformation. The features shown are largely self-explanatory, although I should explain that the green shaded portion represents where the F1 and F2 polypeptides overlap. This shows one of the protomers:
- There was no challenge to the accuracy of this schematic or Professor Weissenhorn's evidence in relation to it. Profession Wilkinson accepted it was an accurate schematic representation.
- In cross-examination it was suggested to Professor Wilkinson that the way in which the fusion peptide is being prevented from accessing the membrane in the Pfizer construct is not substantially the same as the way in which the fusion peptide is being restricted or persuaded not to access the membrane in the claims of either of the Patents. His responses were that 'the underlying chemistry is different' and 'in terms of the chemistry, it has been done differently, yes.' In closing, GSK sought to dismiss his answers as irrelevant as being at a level of generality different to that of the claims. However, the 'chemistry' is, in effect, the mechanism of the stabilisation the subject of the claims.
- GSK's position is that the inventive core is grounded in the immunological significance of the invention. The Patents state that the invention "concerns the field of immunology" (paragraph [0001]), state that the antigens are useful as vaccines, and describe immunological work in animal models to establish the utility of the constructs. The inventive cores of claim 1 of EP 258 and of EP 710 are the use of an RSV antigen, in which the prefusion conformation of the F protein is stabilised, as an immunogen.
Analysis
- The relevant 'result' in the context of EP258 is an antigen with the functional features of claim 1 which is stabilised in the prefusion conformation. The 'way' that the stabilisation is achieved is by the addition of the heterologous trimerization domain positioned C-terminal to the F1 domain.
- In this regard, I have no doubt that GSK's argument is at an inappropriately high level of generality. In effect, their argument amounts to claiming any construct in which the prefusion conformation of the F protein is stabilised. That, however, still requires scrutiny of Pfizer's argument.
- Question 1 is whether the non-single polypeptide of Pfizer's product achieves substantially the same result in substantially the same way as the claimed polypeptide. Pfizer say no, because it is not a single polypeptide that is stabilised by a trimerization domain, they say it is two polypeptides that are locked in place by that extra disulphide bond, and other features which include a trimerization domain. Pfizer argue that the stabilisation in Pfizer's product is not achieved in substantially the same way.
- On this point, I consider it is necessary to keep in mind the point that the evidence to the effect that the degree of stabilisation provided by the addition of the trimerization domain was modest, with the corresponding effect on the construction of 'stabilizes'.
- The further point which I consider should be noted is that, if the RSVPreF construct is held to infringe through the doctrine of equivalents because it achieves the result of stabilization in substantially the same way as in claim 1 of EP258, the scope of the claim would be broad: any F protein stabilised in the prefusion conformation which happened to include a trimerization domain would be likely to infringe, regardless of the influence of other stabilisation features. I do not consider it is necessary to strain to extend the scope of this claim when the patentee had a free choice to specify which stabilisation strategy or strategies to include in the claim. Furthermore, it may be expected that the patentee has, via divisionals, claimed other stabilisation strategies.
- Overall, I conclude that, due to the combination of the RSVPreF construct having two polypeptides locked in place via the non-native disulphide bond, that combination does not achieve the result of stabilisation in substantially the same way as in claim 1 of EP258, not least because the degree of stabilisation achieved is materially different to that achieved solely by the addition of a trimerization domain. As Professor Wilkinson said, the chemistry (which I interpret as the mechanism of stabilisation) is different. So, the answer to question 1 is no.
- In case I am wrong about that, I go on to consider question 2. This is a case where the fact that the skilled team knows that the variant achieves substantially the same result as the invention makes no difference because that would be clear to the skilled team. However, I conclude that it would not be obvious to the team that the variant achieves that result in substantially the same way as the invention. They would regard the use of two polypeptides locked together by the non-native disulphide bond as a different way, notwithstanding the presence of the trimerization domain as well as other stabilising features.
- In case I am wrong in my conclusions on both questions 1 and 2, I go on to address question 3.
- For this purpose, I must reset and assume that the RSVPreF does achieve substantially the same result in substantially the same way and that was obvious to the skilled reader. However, even in that scenario, the skilled reader would have noticed the definition of 'polypeptide' in the specification and the deliberate limitation by the use of that defined term in the claim.
- Pfizer contended that the skilled reader would understand the use of a single polypeptide to be fundamental to all of the stabilizing teaching of the Patents and so for strict compliance to be intended, even if the first two questions were answered in the affirmative. As far as I can see, the Patents do not say anything explicit about the importance of a single polypeptide, although it could be said to be implicit from the fact that the Patents only address that situation.
- I have found this question 3 more difficult and the considerations seem to me to be very evenly balanced. The balance comes down, in my view, in favour of an affirmative answer, not least because of the considerations I mentioned in the last two sentences of [535] above.
- For the purposes of Actavis question 1, GSK identified two different inventive concepts:
i) the production of a recombinant RSV antigen modified to stabilise the prefusion form; or in the alternative
ii) the use of an RSV antigen, in which the prefusion conformation of the F protein is stabilised, as an immunogen, wherein stabilisation is achieved by a modification to prevent or reduce membrane accessibility to the N-terminal part of the fusion peptide.
- By contrast, Pfizer contends that the inventive concept is the modification of an RSV F protein polypeptide by the removal of furin cleavage sites and addition of a trimerization domain.
- Neither of GSK's inventive concepts is appropriate. Both are pitched at far too high a level of generality. Pfizer's is pitched far nearer to the level of generality of the claim.
- Pfizer contended that in EP710 the single polypeptide is stabilised in a manner which reduces the likelihood of triggering the postfusion form first, by avoiding the proteolysis through having no furin cleavage sites between the F1 and F2 domains and second by addition of the trimerization domain. Thus, Pfizer submitted, the purpose of the removal of the furin cleavage sites in EP710 was to prevent the single polypeptide claimed from being cleaved.
- In their written closing, Pfizer submitted that in the RSVPreF, the furin cleavage sites are retained. I confess I did not understand that submission because, as the PPD explains at [6], the mature protomer is formed by excision of the pep27 domain at the dual furin cleavage sites. The sequence information in the PPD shows the excision of pep27 which ends with the sequence of the second furin cleavage site RKRR. The F2 domain is left at its C-terminal with the sequence RARR, which is the sequence indicating the first furin cleavage site, but the cleavage occurs immediately after that sequence. Although there was no evidence specifically to this effect, it seems logical that once a furin cleavage has occurred, the sequence is then rendered ineffective and spent - there is nothing to cleave after RARR in the F2 domain. The final point is that there was no evidence to support Pfizer's submission, so I leave it out of account.
Analysis
- Claim 1 of EP710 claims an antigen defined in structural terms. The structural feature which runs throughout is the single polypeptide. The last structural requirement is that the (single) polypeptide comprises a trimerization domain positioned C-terminal to the F1 domain. RSVPreF has a trimerization domain positioned C-terminal to the F1 domain, albeit the F1 and F2 domains comprise two polypeptides.
- As for the other structural requirement, in one sense, it is true that there are no furin cleavage sites between the F1 and F2 domains because they already comprise two polypeptides. However, isolating that requirement of the claim ignores the fact that those two domains must be of a single polypeptide. In the RSVPreF, the absence of the furin cleavage sites does not prevent cleavage of the single polypeptide, because F1 and F2 are already separated into distinct polypeptides.
- As explained in [0067], 'Removal of one or both of the furin cleavage sites is predicted to prevent membrane accessibility to the N-terminal part of the fusion peptide, stabilizing the prefusion state.'
- In the RSVPreF, the fact that the F1 and F2 domains are separate polypeptides does not expose the fusion peptide. One cannot avoid the fact that in the RSVPreF membrane accessibility to the fusion peptide is prevented by the F1 and F2 domains being locked in place by the non-native disulphide bond.
- For these reasons, I conclude that for the RSVPreF construct, both the result and the way are different. The chemistry is very different. The RSVPreF construct does not achieve substantially the same result (the structural features of claim 1 of EP710), and the way the structure of the RSVPreF is achieved is also different. So the answer to question 1 is no.
- Question 2: Again, the second question does not arise in the light of the answer to question 1. In this instance I find it difficult to address question 2 on the alternative basis (i.e. if I am wrong on question 1) because of my finding that both the way and the result are different.
- Question 3: In view of the fact that claim 1 of EP710 is expressed in structural terms, and for the reasons explained when addressing question 1, in my view the skilled reader would conclude that the patentee intended strict compliance with those structural elements to be an essential requirement of the invention.
- Pfizer relied on WO456 (International application PCT/US2008/066223) as a novelty only citation as at the claimed Priority Date per s.2(3) Patents Act, and as an obviousness citation as at the filing date (23 December 2008). Pfizer challenged the entitlement to priority of EP 258 and EP 710 on formal grounds. The challenge to priority turns on GSK's failure to establish it was the successor to the right to claim priority from the priority documents at the filing date. It requires consideration of Belgian law.
- It is appropriate to address priority here even though it is a complete diversion away from the technical issues. As indicated above, the challenge turns entirely on an issue of Belgian law which has not been decided by any Belgian Court.
- The four inventors, who devised the invention of the Patents during their employment at the Defendants, filed two US patent applications (being US 524 and US 206). Those same four inventors together as joint applicants with the Defendants subsequently filed a PCT application (being WO 456) which claims priority from the two US patent applications. The PCT application entered the national/regional phase at the EPO and subsequently gave rise to EP 710 and EP 258.
- The four inventors are Mr Blais and Mr Rheault (who were employed by the Second Defendant in Canada) and Mr Baudoux and Mr Ruelle (who were employed by the First Defendant in Belgium). Pfizer accepts that the Canadian inventors had assigned the relevant rights to the Second Defendant as at the date of filing of WO 456. Pfizer also accepts (and, indeed, positively avers in evidence) that Mr Baudoux's relevant rights were transferred in time. However, Pfizer denies that Mr Ruelle assigned his relevant rights to the First Defendant before WO 456 was filed.
- The issue is whether GSK are entitled to claim priority from US 524 and US 206.
- The law on priority was considered in KCI Licensing v Smith & Nephew [2010] EWHC 1487 (Pat), where Arnold J accepted an analysis based on common law principles distinguishing equitable and legal title to property. If the relevant local law meant that the equitable or beneficial title to the priority right was in the hands of the person making the priority claim in the international application, that was held to be good enough even though that person did not then hold the legal title under the local law and could only perfect their title after the event.
- The judgment of Birss J in Accord Healthcare Ltd v Research Corporation Technologies, Inc (Rev 1) [2017] EWHC 2711 (Pat) refers to this at [68]:
'The critical passage in KCI is as follows. Arnold J had held that on its true construction the relevant agreement there did convey the legal title to the applicant but he went on to hold that even if that was wrong, the agreement was effective to transfer the entire beneficial interest. The applicant had an enforceable legal right to call for a conveyance of the bare legal title and that made the applicant the "successor in title" for the purposes of a claim to priority under Article 87(1) of the EPC and Article 4(A)(1) of the Paris Convention even if KC Inc had not acquired the bare legal title at the relevant date. After referring to a decision of the EPO Case J19/87 Burr-Brown /Assignment [1988] EPOR 350, Arnold J held:
'71. To my mind, this makes sense. Article 4(A) of the Paris Convention and Article 87(1) of the EPC are provisions in international treaties whose operation cannot depend upon the distinction drawn by English law, but not most other laws, between legal and equitable title. When determining whether a person is a "successor in title" for the purposes of the provisions, it must be the substantive rights of that person, and not his compliance with legal formalities, that matter.''
- The legal principles that bear on priority were then summarised by Birss J at [75]:
'I find that the legal principles applicable to priority entitlement are settled at this first instance level. They are:
i) Usually the right to claim priority goes with the right to the invention. That is uncontroversial.
ii) The right to claim priority must be with the person making the patent application in which that right is claimed when they make that claim, i.e. when the application is filed. A later acquisition of that right cannot make good a lack of it on the relevant date. If the right was not in place at the time then the right is lost for all time. That is Edwards v Cook.
iii) But if the local law applicable to rights of the applicant and the patent application at the place and time when it was made allows for a splitting of property rights into legal and equitable interests, then it will be sufficient to establish an entitlement to priority if the applicant holds the entire equitable interest at the relevant date. That is KCI, HTC and FujiFilm and was held in the Court of Appeal in Idenix provisionally to be correct.
iv) A person with a legally enforceable right to call for the assignment of the legal title to a piece of property such as an invention (or a right to claim priority) has the equitable title to that property. When the cases refer to the applicant holding the substantive right and title to the invention, they are referring to this legal/equitable distinction.'
- Pfizer called Professor Buydens to give evidence on Belgian Law. Professor Buydens is a co-head of the IP and IT team at Janson Baugniet, a Belgian law firm, and has been practising in IP litigation since 1992. She has been a professor at the Free University of Brussels since 1995 where she teaches IP and information law. She has also been a Professor of Patent Law at the University of Liège since 2021. Pfizer submitted she was an impressive witness and that her opinions on Belgian law should be given the highest weight. GSK submitted she took unmeritorious points on the key issues. I assess these points below.
- Mr Ronse was called by GSK. He is an attorney who has practised in Belgium for 34 years. He is a partner in the IP and litigation department of law firm Altius, based in Brussels. Pfizer sought to criticise Mr Ronse on the basis that he was a litigator and tended to argue GSK's case rather than answer the points put to him. Accordingly, Pfizer suggested that his evidence should be treated with caution where it was not supported by authority or commentary. Once again, I assess his evidence in the context of the points which were in issue.
- In general terms, both of the Belgian law experts were trying to assist me but, to varying degrees, both occasionally avoided giving a clear answer to a straightforward question, as if reluctant to be pinned down to an answer unhelpful to their instructing party. Nonetheless, I am grateful to both of them for their assistance.
- GSK contended that the following principles of law apply.
i) First, that a claim to priority will be good if the applicant had on the date of filing the application (i) legal title to an invention, or (ii) the substantive rights in an invention where legal formalities were yet to be perfected.
ii) Second, that the assignment of an "invention" will include the right to apply for a patent and necessarily priority rights deriving from any patent application. GSK contends that Article 2 of the 1986 Contract (Mr Ruelle's contract with his employer being a GSK entity) contains an assignment of future rights of any inventions Mr Ruelle made in the course of his employment.
- Art. 2 of the 1986 Contract provided as follows:
'The undersigned party of the second part [i.e., Mr Ruelle] acknowledges that since his activity for s.a. SMITH KLINE-RIT includes, by its very nature, an inventive mission or knowledge of the carrying on of inventive tasks within the company, directly or indirectly, in whole or in part, permanently or intermittently, this confers on any inventions resulting therefrom the status of so-called "service" inventions, regardless of how its activity carried out for the undersigned party of the first part [i.e., GSK] is classified. The undersigned party of the second part [Mr Ruelle] acknowledges accordingly that the contractual remuneration adequately covers the carrying on of his activity, as described, and that the inventions that he may make, in which he may collaborate or of which he may become aware, under no circumstances constitute his property, but that of the undersigned party of the first part [GSK] or the persons designated by it." (emphasis added)
- Against this backdrop, Pfizer pleaded the following three points:
i) That the 1986 Contract does not concern the priority right
ii) The 1986 Contract is a 'promise to contract' (or an option) rather than an assignment of future rights. It then says that this option became limitation-barred.
iii) The PCT request form by which the two Defendants and the four inventors jointly applied for the PCT. Mr Ruelle (and Mr Baudoux) were both designated for "US only".
- Pfizer also took an unpleaded point, identified in their Opening Skeleton. Pfizer alleged there was no evidence of any transfer of rights from Mr Ruelle's employer, SmithKline RIT S.A. to the applicant for the PCT, the First Defendant. Whilst submitting that it was not open to Pfizer take this point, GSK nevertheless submitted they had a good answer to it.
- I address these four points in turn.
Whether the 1986 Contract concerns the priority right
- Neither party identified a Belgian case dealing with a Belgian law contract on the specific issue of whether on an assignment of the invention, that the language in Article 2 encompasses the priority right.
- The evidence provided by Mr Ronse on Belgian law explained that if an inventor assigns to the industrialist both his application for an invention and the right to file in all countries, the assignee may introduce these new applications under the benefit of a priority right, even if the agreement entered into does not contain any express stipulation as to the right of priority.
- Where Mr Ruelle may have devised a number of inventions for GSK during the court of his employment, the rights in those inventions will have been assigned at the time at which they came into being. This means that even if the priority right comes into effect when the patent is filed and after the invention was devised, it remains an assignment of future rights, and so it is still carried with it. Whilst the priority right and the right to file a patent are distinct rights, this does not impact upon this analysis as Article 2 is concerned with the transfer of the invention, and does not differentiate between those various rights that are bundled up with that.
- Pfizer contends that under the combined effect of Articles 2 and 3, there is no need, for the agreement to assign the priority right. GSK contends that this however does not deal with the proper interpretation of the assignment of an invention, and indeed whether or not the contract needed to assign something does not meet the point.
- Parallel to this analysis, under the doctrine of service inventions there is an issue on Belgian law as to whether the default rules of service inventions would provide for that same outcome and transfer the priority rights.
- Article 2 of the 1986 Contract states that any inventions devised by Mr Ruelle during his employment have the status of "service inventions". Mr Ronse gave evidence that service inventions are considered to vest in the employer automatically unless such is contractually excluded by the parties.
- Professor Buydens took two points against this. The first is that the rules of service inventions apply only in the absence of a contract. Mr Ronse however cited three Belgian cases taken from exhibits to Buydens I in which the Belgian court considered the ownership of an invention under the rules of service inventions in conjunction with the contractual position. The second point was that the employee's presumed consent as to the rights which they agreed to transfer has to be interpreted restrictively. She relied on a quotation from Professor Janssens' book which was selective in that the very next paragraph of the cited work confirmed that the agreement to transfer rights to the employer is applicable for service inventions.
- I conclude that, on a proper interpretation, the 1986 Contract does extend to encompass the priority right.
Whether the 1986 contract operates as an assignment of future rights
- The second point is whether the 1986 contract operates as an assignment of future rights, as GSK says, or as a promise to contract, or like an option, as Pfizer says.
- It is common ground between the parties that there is a principle of Belgian law that the pre-eminence of the written contract prohibits an interpretation that is irreconcilable with the wording of the contract.
- GSK rely in particular on the language where it says under no circumstances will the property in the invention be Mr. Ruelle's. If the 1986 agreement acts like an option, there will be circumstances under which Mr. Ruelle owns the property in that invention, at least for the time being until the option is exercised. GSK submitted that that is an interpretation irreconcilable with the wording of the contract and so not to be preferred.
- In determining whether the 1986 Contract concerns the priority right, I consider Pfizer's argument relating to the indeterminacy of the assignee. That is, Article 2 refers to the undersigned party of the first part or persons designated by it. Pfizer says that cannot be an assignment because the assignee is uncertain.
- Article 2 is properly understood as saying that the assignee is GSK, unless and until GSK designate another entity. That is a straightforward and practical interpretation of that article.
- It can be understood that was GSK's intention for the provision to operate in that way given that over the course of their employment Mr. Ruelle may have collaborated with other entities, and it might be a useful provision to allow it to be assigned to another designated party, but that does not affect the position that GSK is the assignee.
- GSK did have a secondary position in relation to this - if they are wrong and Article 2 is an option, then GSK contend that is still sufficient to render GSK entitled to priority and meets the requirements under section 5 of the Act.
- Belgian law does not have a legal/equitable distinction, but on Pfizer's analysis, the promise to contract is a legally enforceable right to call for the assignment of the legal title.
- Pfizer's position is that the promise to contract had become limitation barred, with which GSK disagrees. The essential point between the parties is whether or not the time for limitation runs from when the invention was devised or whether it runs from an earlier date, like when the contract was executed. That is GSK's secondary position. GSK's primary position is that this was a present assignment of future rights.
US designation
- This pertained to the request form for the PCT application that became WO456 and gave rise to the patents. On this PCT application Mr. Ruelle was designated for the US, Pfizer therefore argue he must have retained the US priority right, because that is needed for him to claim priority in the US. That premise involves a matter of US law as to whether the position was such that Mr. Ruelle could or could not claim priority for that application.
- Pfizer neither pleaded nor led any evidence on US law. In any event, Mr. Ronse explained the concept of name lending, like agency. Pfizer has not suggested that, from the US perspective, name lending or agency analysis is the wrong one.
- The second entry on the PCT request, Mr Baudoux, is in the same position as Mr. Ruelle, in that he is designated for the US only. GSK say this means Mr. Baudoux had transferred his priority right to GSK and yet he is shown in the same position as Mr. Ruelle on the PCT request form.
Were the relevant rights assigned to the First Defendant
- This point unfortunately requires an analysis of the pleadings.
- When Pfizer issued their claim for revocation, they put GSK to proof of any claim to priority, alleging that the applicants for the PCT were not the successors in title to the relevant rights when the PCT was filed. GSK's Defence pleaded out the chain of title. In their Reply, Pfizer admitted that Mr Ruelle entered into the 1986 Contract with SmithKline RIT S.A. but denied in general terms that the agreement "had the effect of assigning or otherwise transferring to Smith Kline-RIT S.A. or agreeing that Smith Kline-RIT S.A. was or was to be the owner of any right of Jean-Louis Ruelle to claim priority". It entered a general non-admission against GSK's case. As GSK submitted, no point about the chain of title between GSK entities was identified. Nonetheless, Pfizer were right to point out that the burden of proving entitlement to priority lay on GSK.
- Following a request by GSK for further information, the parties agreed that they would enter pleadings on priority and Belgian law, with Pfizer pleading first. The CMC order accordingly provided:
"6. On or before 2 December 2022, the Claimant shall file and serve a Statement of Case on Entitlement to Claim Priority and Belgian Law setting out all facts and matters relied on in support of its case that WO456 is not entitled to priority.
7. On or before 20 January 2023, the Defendants shall file and serve a Statement of Case on Entitlement to Claim Priority and Belgian Law in reply."
- GSK pointed out, correctly, that:
i) Pfizer's pleading on priority and Belgian law raised no issue about the intra-GSK assignment, yet the CMC Order required Pfizer to plead the point if it was going to be taken.
ii) The point featured nowhere in Pfizer's evidence on priority and Belgian law. Indeed, Buydens I paragraph 100 stated: "I understand that SmithKline RIT SA later became GlaxoSmithKline Biologicals S.A. (who I hereafter refer to as "GSK") as evidenced by document D39".
iii) The point emerged for the first time in Pfizer's Opening Skeleton for Trial.
- GSK contended that if the point had been pleaded, they could have provided further disclosure and/or fact evidence about the intra-GCK chain of title, with attendant expert evidence as necessary. Fortuitously, GSK had provided, in its initial disclosure, documents notifying Mr Ruelle of the transfer of his employment between the relevant GSK entities. These documents were also the subject of a CEA Notice.
- My conclusions are as follows:
i) Under the terms of the CMC Order, if Pfizer wished to take this point, it had to be pleaded, but it wasn't.
ii) GSK was therefore deprived of the opportunity to investigate whether further disclosure or evidence could assist.
iii) Nonetheless:
a) the letter of notification to M. Ruelle dated 25 January 1989, notifying him of the transfer of his employment from SmithKline RIT S.A. to SmithKline Biologicals S.A., together with
b) the letter of notification to M. Ruelle dated 25 September 1990, notifying him of the transfer of his employment from SmithKline Biologicals S.A. to SmithKline Beecham Biologicals S.A., plus
c) the statement in the Belgian Official Gazette of 30 March 2001 of the change of name of SmithKline Beecham Biologicals S.A. to GlaxoSmithKline Biologicals S.A.
provide sufficient evidence of novation of M. Ruelle's contract of employment from SmithKline RIT S.A. through to the First Defendant.
iv) In view of M. Ruelle's apparent long-standing employment in the GSK group and the period of many years between September 1990 and the filing of US524, US206 and WO456, I consider it is safe to infer that when the invention(s) were made, M. Ruelle was employed by GlaxoSmithKline Biologicals S.A. under the terms of the 1986 Contract as novated to that entity.
- Accordingly, I reject this unmeritorious fourth point.
- Accordingly, in light of the above, GSK was entitled to claim priority deriving from Mr Ruelle's rights in the two US Patent applications, US 16524 P and US 56206 P at the filing date of WO456.
- It was in the light of this conclusion that I did not feel it necessary to address the issue of obviousness over WO456, which would only have become applicable if priority had been lost.
- To anticipate, the prior art disclosure must 'plant the flag' i.e. it must be a clear and unambiguous disclosure of all of the features of the claim.
- I have also reminded myself of certain dicta made by Pumfrey J. in Research in Motion v Inpro [2006] EWHC 70 (Pat):
i) First, at [111]: 'A claim lacks novelty if it covers something that formed part of the state of the art at the priority date'.
ii) Second, at [112]: 'The teaching of the specification, once construed, is a pure question of fact, as is what the skilled man would do with that teaching without the exercise of inventive ingenuity.'
iii) Third, at [128]: 'As ever, the question is what is explicitly disclosed and what also is necessarily implicit in the teaching. The skilled man must be taken to read documents in an intelligent way, seeking to find what is disclosed as a matter of substance.'
- WO456 is a patent application entitled "Methods and compositions related to viral fusion proteins" which was published on 18 December 2008. Mark Peeples is listed as an inventor. It is a long document and it is only necessary to refer to selected parts, as follows.
- In the Background section at paragraphs [0003] to [0007], WO456 provides an overview of the Paramyxoviridae family and briefly describes the clinical aspects of RSV-mediated disease. Paragraph [0006] states that there is currently no vaccine or specific treatments against RSV, and that the failure in developing a vaccine has led to renewed interest in the pathogenesis of the disease.
- The 'Summary of The Invention' is at paragraphs [0008] to [0013]. [0008] says
'Provided herein is a pre-triggered soluble fusion (F) protein of a virus in the paramyxovirus family, wherein the soluble fusion protein lacks a transmembrane domain and a cytoplasmic tail domain and includes a CRAC1 domain. The soluble fusion protein is in a pre-triggered conformation and can be triggered when exposed to a triggering event.'
- After the brief description of the figures and the start of the Detailed Description, I can pick it up in the Embodiments section, where, at [0077], WO456 states that:
"Contemplated herein is an isolated soluble fusion (sF) protein of a member of the paramyxovirus family in its pre-triggered form".
- A "soluble" F protein is defined in paragraph [0079] as a truncated fusion protein that is not membrane-bound, so lacks the transmembrane and cytoplasmic tail domains. In some embodiments, the pre-triggered sF protein also lacks the pep27 region.
- Paragraph [0085] describes the use of detection tags for identification and purification of the constructs. It gives range of examples, including the 6His and FLAG tags, which are used in the embodiments.
- Paragraphs [0086] and [0087] describe the use of C-terminal "clamp" in certain embodiments in order to stabilize the pre-triggered form. It is worth reproducing those sections in full here:
"[0086] In some embodiments, the sF protein contains a C terminal "clamp" to hold the C terminus of the protein in position. The clamp holds the C termini of the three monomers in the molecule together, preventing them from separating or moving upward and triggering the molecule. In one example, the C terminal clamp is a trimerization domain, such as GCNt. The sF protein with the GCNt clamp that we produced, sMP340-A, is secreted efficiently from transfected cells but it is not recognized efficiently by MAbs against the F protein, may be partially aggregated, and is not triggered by treatment at 50C for one hour. Minor modifications to this construct, however, will likely result in a pre-triggered sF protein. Those modifications include removal of the glycine that we had inserted between the sF protein C terminus and the GCNt clamp to add flexibility, removal of residues or insertion of residues such as alanine, that will not disturb the helical nature of this region but which can bring the HR2 helix and the GCNt helix into phase with each other. In another example, the clamp contains a trimerization domain comprising two cysteines that will covalently link the three monomers. In this example, two amino acids at or near the C terminus of the HR2 helix in each soluble F protein monomer are replaced with two cysteines. The cysteines are either consecutive or have one or more amino acids separating them. The 6 cysteines in the trimer will form 3 disulfide bonds, linking the C termini of the three monomers.
[0087] For example, the sF protein stabilized at its C terminus by either the addition of a GCNt clamp or cysteines are useful tools for assessing the first step of triggering, i.e., unfolding of the HR1 domain, without the second step of forming the 6-helix bundle. Because the HR2 helices are linked in this protein, they will not be able to fit into the grooves provided by the HR1 trimer to produce the 6-helix bundle. On the other hand, the sF protein without the cysteines will be able to perform both unfolding of the HR1 domain and formation of the 6-helix bundle because its C terminus is not cross-linked to the other monomers in the trimer. So, the clamp or the Cys linkage would probably stabilize the sF protein making it easier to store and to use since more of it would remain in the pre-triggered form. For example, SC-2 begins to decay as soon as it is made, with a t1/2 of about 3 weeks".
- WO456 states that several strategies are available to produce and maintain and/or stabilize isolated F proteins in the pre-triggered state. It goes on to state (at the end of [0088]) that:
"As described above, the sF protein may also be physically stabilized by adding a GCNt segment to clamp the C terminus, or by adding cysteines that will cross-link the trimer C termini."
- Paragraph [0096] describes other embodiments in which pep27 (aa 110-136) is removed or replaced with alanine and glycines "without destroying the function of the F protein".
- The section at paragraphs [00102] to [00114] describes general methods of producing the pre-triggered, soluble F proteins. It covers techniques such as the use of suitable promoters, expression of the proteins in mammalian cells, such as CHO cells and the use of transfection plasmids.
- The sections starting at para [00115] and [00140] describe using computational models of RSV F in the pre- and post-triggered form to design or screen for potential anti-viral agents.
Example 1: computer modelling of the RSV F Protein ([00189])
- The authors state that they modelled the pre-triggered and post-triggered forms of the RSV sF protein based on the X-ray crystallographic structures of the PIV5 and PIV3 sF structures, citing the two Yin papers and referring to Figures 2-6 (described above).
- The models were generated by "threading" the RSV F sequence onto the C chain of the PIV5 or PIV3 crystal structures. SwissModel was used to independently generate structures for the F1 and F2 strands.
Example 2: methods for generating soluble RSV F proteins ([00191]
- This section describes the production and characterization of three soluble RSV F constructs. One of them, designated sMP340-A, contains a GCNt domain at the C-terminal end of the fusion protein to clamp that end of the molecule and stabilize it. See paragraph [00195], which states:
"The three versions of the RSV sF protein (cartoon in Fig. 10; sequences in Fig.11) were constructed from MP340 by replacing the transmembrane and cytoplasmic domain of the F protein gene: 1) with a FLAG tag followed by a 6-histidine (6HIS) tag (SC-2); 2) and the last two amino acids of the HR2 helix (523 and 524) with two cysteine residues to allow the C terminus of the F sequence in the trimer to covalently link the monomers, followed by a FLAG tag followed by a 6HIS tag (HC-1); and 3) with a TEV protease cleavage site followed by a GCNt trimerization domain followed by a FLAG tag followed by a Factor Xa cleavage site followed by a 6HIS tag (sMP340-A). These novel sequences replacing the C terminus of the RSV F protein were designed to purify the sF protein released into the medium of transfected cells (6HIS tag or FLAG tag), enable easy detection of the sF proteins (6HIS tag or FLAG tag), or to clamp this end of the molecule to stabilize it (covalently with cysteines or non-covalently with the GCNt trimerization domain)."
- The structures of these RSV F protein constructs are also shown in Figure 10 of WO456, included below. The schematic shows that SC-2 comprises a wild type sequence with the transmembrane domain removed to allow the production of soluble protein. The HC-1 construct includes 2 additional cysteine resides at its C-terminal end, described in Fig 10 as "2 Cys". The sMP340-A construct includes a GCNt domain at the C-terminal end, which is described here as a "self-trimerizing clamp", positioned between additional Factor Xa and TEV (tobacco etch virus) protease sites. As shown there are two disulphide bonds between the upper F2 and lower F1 segments (and similarly for the other depictions).
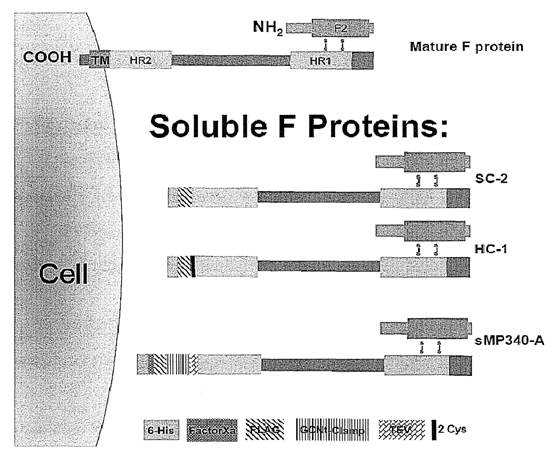
- The section goes on to describe the production and expression of the three proteins in human embryonic kidney 293T cells. After expression, the proteins were stored at -20 degrees C for later characterization. As the recombinant proteins had their furin cleavage sites intact they were fully cleaved.
- Paragraphs [00203] to [00214] describe various experiments to test whether the recombinant RSV F constructs were in the pre-triggered or post-triggered form. In the first experiment, described in [00203] and Fig 14, the constructs were analysed by ultracentrifugation through linear sucrose gradients. The authors described the results of that experiment as follows:
"In our initial experiments, both SC-2 and sMP340-A migrated further into the sucrose gradients than expected (Fig. 14), indicating that they were aggregated. We had expected SC-2 to migrate in this manner, indicative of aggregation, but not sMP340-A. We hypothesized that freezing the protein between the time of production and purification and the sucrose gradient might be responsible for the sMP340-A migration indicating aggregation."
- The authors then repeated the experiment again at 4 degrees C (so without freezing the proteins to -20) and again after heating them to 50 degrees C. The results are reported in [00204] to [00208] and shown in Figure 15. WO456 states that the sMP340-A protein did not stay at the top of the sucrose gradient, but neither did it move further down after heat treatment at 50 degrees "suggesting that this protein is not in the pre-triggered form to begin with and could not be triggered". WO456 provides a possible explanation for why they obtained that result: "It is possible that the GCNt trimerization domain distorts the RSV sF protein. However, it is also possible that the GCNt domain that we added to the sF sequence was not in the proper phase with the HR2 domain, resulting in a distorted protein.'
- The SC-2 protein remained near the top of the sucrose gradient, which the patent says was "indicative of the pre-triggered form". After heating to 50 degrees the sF protein migrated further into the gradient, indicating that it had aggregated and therefore been triggered to the post-triggered conformation.
- At paragraph [00207], WO456 states that "The HC-1 sF protein behaved just like the SC-2 sF protein (Fig. 15), demonstrating that it, too, is a pre-triggered sF protein that can be triggered by heat".
- Under the next heading 'Confirming the pre-triggered state by reactivity with neutralizing antibodies':
"According to our hypothesis, any mouse monoclonal antibody (MAb) against the F protein that neutralizes RSV infectivity in cell culture would bind to the virion form of the F protein and probably to the pre-triggered form of the F protein. If the SC-2 or sMP340- A protein represents the pre-triggered form of the sF protein, neutralizing MAbs should recognize it."
- The authors tested binding of the constructs to a panel of 11 neutralizing monoclonal antibodies that were available to them. The patent states at paragraph [00209] that:
"All 11 of these MAbs immunoprecipitated the SC-2 sF protein efficiently (Fig. 16, "-" lanes) suggesting that this sF protein is in the native F protein conformation. The same 11 MAbs did not immunoprecipitate the sMP340-A sF protein efficiently, suggesting that sMP340-A may not be in the native conformation."
- The authors wanted to test the possibility that if heat caused triggering of the protein then it should also cause the loss of one or more of the epitopes recognised by the antibodies. The logic was that if the neutralizing antibodies bind predominately to the pre-fusion or pre-triggered form of the protein, when the protein switches to the post triggered form antibody binding will reduce. To test this, they heated both the SC-2 and sMP340-A constructs to 50 degrees for an hour before immunoprecipitating them with the panel of 11 neutralizing antibodies In Figure 16, the "–" lane indicates before heating and the "+" lane indicated after heating. The results of the antibody binding experiments are shown in Figure 16 and described as follows, in [00213]:
"The heated SC-2 sF protein lost its ability to be recognized efficiently by all 11 of the Mabs (Fig. 16, "+" lanes), indicating that heating had caused major conformational changes in the SC-2 sF protein, consistent with it being triggered by the heat treatment. Heating the sMP340-A sF protein had no effect on MAb binding (Fig. 16 "+" lanes), indicating that sMP340-A is not triggered by mild heat."
- WO 456 is said by Pfizer to anticipate claim 1 of EP 258 only. If priority is lost, Pfizer relies on it as an obviousness citation against all claims of EP258 and EP710 in issue.
- Pfizer's case is that all of the features of claim 1 are met. WO456 discloses a recombinant RSV antigen, sMP340-A, with all of the structural features of claim 1.
- Dr Taylor's view was that the key difference between sMP340-A and claim 1 of EP 258 is that sMP340-A is not in the pre-triggered (prefusion) conformation. Professor Wilkinson's view in relation to the first experiment in which the constructs were frozen was that sMP340-A was aggregated and that the freeze/thaw cycle is "associated with denaturation and/or aggregation". Professor Weissenhorn agreed that the sMP340-A construct is unlikely to be in the prefusion form.
- In relation to the second experiment (described in paragraph [00204]), the authors report that sMP340-A is not in the pre-triggered form but was likely distorted by the addition of the GCNt domain not being in the proper phase with the HRB domain. Professor Wilkinson explained that the distortion was probably due to the inclusion of a TEV protease site between the C-terminus of the F protein and the GCNt domain, the TEV protease site being a 22-amino acid residue that does not possess obvious heptad repeat character. In cross-examination, Professor Weissenhorn agreed and reasoned that "the [TEV protease] sequence has no propensity to form a coiled coil".
- Given that the modifications made by the authors of WO 456 to produce sMP340-A would not have stabilised the prefusion form, WO 456 does not anticipate claim 1 of EP 258.
- In light of the other findings I make on validity of the Patents in this Judgment, I do not consider it is necessary to address the alternative case of obviousness over WO 456 in case I am wrong on the Belgian law issue.
- The parties seemed to be in agreement that the issues of obviousness raised in this case required an application of standard and well-known principles.
- For obviousness, the correct legal approach is that summarised in Actavis v ICOS [2019] UKSC 15 at [52]-[73] per Lord Hodge, referring to the structured approach in Pozzoli v BDMO [2007] EWCA Civ 588 at [14]-[23] per Jacob LJ and citing Kitchin J. in his well-known passage from Generics v Lundbeck [2007] EWHC 1040 (Pat) at [74].
- Pfizer relies on the remaining prior art:
i) The abstract entitled "Structures of the pre- and post-entry paramyxovirus F protein: implications for RSV vaccine and therapeutic development", Jardetzky T et al., from the abstract booklet from the Sixth International RSV Symposium (the "RSV Symposium"), held in Florida, USA on 25 to 28 October 2007 (the "Jardetzky Abstract");
ii) The public presentation "Structures of the pre- and post-entry paramyxovirus F protein: implications for RSV vaccine and therapeutic development" delivered by Prof Jardetzky at the RSV Symposium (the "Jardetzky Slides") (it is common ground that this would be read together with the Jardetzky Abstract);
iii) The oral disclosure of Prof Jardetzky which accompanied his slides in the Jardetzky Presentation (the "Oral Disclosure"). The nature of the Oral Disclosure is in dispute and is the subject of Prof Jardetzky's evidence;
iv) "Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation", Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS., Nature 2006 Jan 5; 439(7072):38-44 ("Yin"); and
v) The abstract entitled "Generation of Soluble Respiratory Syncytial Virus Fusion Glycoprotein" contained within the programme booklet from the 26th Annual Meeting of the American Society for Virology held at Oregon State University on 14 to 18 July 2007 (the "ASV Abstract");
- This is an abstract entitled "Structures of the pre- and post-entry paramyxovirus F protein: implications for RSV vaccine and therapeutic development" which was submitted for the 6th International RSV 2007 Symposium which took place in San Marco Island, Florida on 25 to 28 October 2007. It is a short document with only two substantive paragraphs.
- The International RSV Symposia happens roughly every three years. Both Dr Taylor and Dr Johnson attended the conference but have differing recollections as to the Jardetzky Abstract.
- The first paragraph of the abstract explains that the Paramyxoviridae are enveloped viruses and provides examples of family members including RSV and parainfluenza viruses. It notes that like other enveloped viruses, the paramyxoviruses require fusion of the viral and cellular membranes to enter a host cell. The abstract then explains that the F protein of paramyxoviruses catalyzes 'this membrane merger step' by initially folding to a metastable conformation and subsequently refolding during this process.
- The second paragraph explains that the authors determined the crystal structures of two paramyxovirus F proteins in the pre- and post-fusion conformations. The authors note that the post-fusion conformation of PIV3 F exhibits a "prototypical 6-helix bundle". The structure of PIV5 F pre-fusion conformation was determined 'after stabilizing the metastable state by adding a C-terminal trimerization domain.' Major conformational differences were observed between the pre- and post-entry F structures, involving transformations in secondary and tertiary structure.
- In the final sentence of the abstract, the authors explain that they generated models of RSV F protein and, based on biochemical evidence, they deduce that RSV F undergoes similar conformational changes to PIV3 and PIV5 F. The authors conclude that this has "important implications for RSV vaccine and therapeutic development".
- There had been a dispute between the parties as to which version of the slides were presented. This dispute is not material because, in any event, all of the slides relied on are present in both versions. In both sets of slides the presentation of Prof Jardetzky's group's key structural studies with PIV5 and RSV F prefusion structural modelling is the same, and the Summary & Conclusions slide is the same. From the evidence presented, I agree that the Jardetzky Slides were presented at the RSV Symposium and are thus available as prior art.
- The slides are entitled "Structures of the pre- and post-entry paramyxovirus F protein: implications for RSV vaccines and therapeutic development".
- The slides were presented by Professor Jardetzky at the 6th International RSV Symposia held at Marco Island, Florida on 25 to 28 October 2007 in combination with the abstract of his presentation.
- The second slide provides examples of members of the paramyxovirus family, including RSV and parainfluenza viruses and refers to the two key proteins involved in virus entry (F and G protein).
- On the third slide, schematic diagrams of PIV3 and SV5 constructs are shown. The SV5 construct includes the addition of a C-terminal GCN trimerization domain. The next 4 slides (slides 4-7) present structural diagrams of the F proteins of PIV3 and SV5. The SV5 F protein (described as "F-GCNt") is in the prefusion conformation and the PIV3 F protein is in the postfusion conformation (described as "solF0"). Slide 6 shows the prefusion PIV5 protein structure and postfusion PIV3 protein structure side-by-side for comparison. The image reveals that the prefusion and postfusion conformations are strikingly different. Slide 7 presents a model for F-mediated membrane fusion based on the crystal structures of F-GCNt and solF0.
- Slide 8 lists four unique features of RSV F, specifically that RSV F (i) does not require attachment protein G for viable virus entry, (ii) contains a large insertion upstream of the fusion peptide, (iii) utilizes two furin-like cleavage sites for activation, and (iv) contains an apparent deletion within the fusion peptide. Slide 9 shows the fusion peptide region of PIV5. Slides 10-12 present a computational model of RSV based on the sequences and crystal structure of PIV5. A note on slide 11 states that overall the model is compatible with the SV5 F prefusion structure.
- Slide 14, the "Summary & Conclusions" contains four bullet points. The first bullet point concludes that the F protein can "fold to two very different conformations associated with its membrane fusion function".
- Pfizer also relies on matter which was made available to the public and thus formed part of the state of the art by way of oral disclosure from Prof Jardetzky, given during his presentation of the Jardetzky Slides at the RSV Symposium, in particular:
i) that a heterologous trimerization domain had been used to stabilize the prefusion structure/form of the SV5/PIV5 F protein, a member of the paramyxovirus family;
ii) that information concerning the structures and stabilization of other paramyxovirus F proteins (including in particular HPIV3 and SV5/PIV5) would be important for RSV vaccine and/or therapeutic development;
iii) that the stable prefusion conformation of the RSV F glycoprotein could be highly similar to the SV5/PIV5 prefusion structure, in particular because the RSV F sequence appeared compatible with the SV5/PIV5 F structure and that the major sequence differences could be accommodated in the structure;
iv) that the prefusion conformation of the RSV F glycoprotein would be very useful for developing a vaccine antigen;
v) that the prefusion conformation of the RSV F glycoprotein stabilized with a heterologous C-terminal trimerization domain would be very useful for developing a vaccine antigen;
vi) that the pre- and post-fusion forms of the RSV F protein are distinct and that adding a heterologous C-terminal trimerization domain allows the metastable prefusion form to be stabilized, that stabilizing the prefusion form would be important for developing better vaccine immunogens as that would better represent the form of the protein on the infectious virus, and that parallels can be drawn between RSV and SV5/PIV5;
vii) that the prefusion form of F proteins would have binding sites for antibodies and potentially for small molecules that would be unique and absent in the postfusion form, given the dramatic conformational changes that occur in the protein between pre- and postfusion forms; and
viii) that expressing soluble F proteins without the C-terminal trimerization domain yields the postfusion conformation.
- GSK challenged Professor Jardetzky's evidence on oral disclosure, suggesting it was a reconstruction tainted by hindsight.
- I disagree. Professor Jardetzky gave his evidence clearly and carefully. He was making efforts to recall events from 2007, so naturally he could not have total recall and there were certain matters he recalled and some he did not. He was clear about the strengths and weaknesses of his memory but was firm that he recalled the points conveyed in his presentation, if not the exact words. In this regard, his evidence had a reasonably firm foundation because Professor Jardetzky had given many presentations over the years and evidently had a well-developed practice of using points on his slides as prompts for his oral presentation. There was no suggestion that any of the points set out above as orally disclosed were in any way inconsistent with the content of the Slides. Overall, I found his evidence convincing. In particular, I do not accept GSK's criticisms that his evidence had been influenced or improved by his involvement in an EPO Opposition between these parties.
- Yin reports the crystal structure of the parainfluenza virus 5 F (PIV5) protein stabilized in its prefusion conformation and presents a model of membrane fusion by paramyxoviruses. The protein was stabilized in its prefusion conformation by the addition of a C-terminal trimerization domain.
- The paper compares the crystallized prefusion structure of the PIV5 F protein with a previously reported crystalized postfusion structure of the PIV3 F protein (a closely related virus to PIV5). In the abstract, the authors describe the conformational differences between the prefusion and postfusion states as profound, involving transformations in secondary and tertiary structure. The abstract concludes with this sentence:
'The positions and structural transitions of key parts of the fusion machinery, including the hydrophobic fusion peptide and two helical heptad repeat regions, clarify the mechanism of membrane fusion mediated by the F protein.'
- The first paragraph of the paper introduces the Paramyxoviridae virus family with an explanation that they are enveloped viruses that include, among others, RSV, PIV5, human parainfluenza viruses 1–4 (hPIV), and NDV. The authors then summarize the viral fusion process, noting that paramyxoviruses, like other enveloped viruses such as influenza and HIV, require fusion of the viral and cellular membranes to enter the host cell. The authors explain that two viral glycoproteins are key to this process: an attachment protein and a more conserved fusion protein (G and F, respectively, in the case of RSV). They go on to explain that F is thought to drive membrane fusion by coupling irreversible protein refolding to membrane juxtaposition. It achieves this by initially folding into a metastable prefusion form that then undergoes distinct conformational change to a lower energy state, the postfusion form. They explain that F assembles into homotrimers that are proteolytically cleaved monomers, priming the protein for membrane fusion (like other class I viral fusion proteins such as those of influenza haemagglutinin, HIV Env, Ebola virus GP, and SARS coronavirus S). Following activation, F inserts its fusion peptide into target membranes and refolding occurs, placing the fusion peptides and transmembrane domains in proximity.
- The authors refer to their earlier work reporting on the crystallized postfusion conformation of the PIV3 F protein and note that prior to this paper, it remained unclear to what extent the prefusion and postfusion conformations of the F protein differed and how these were linked to membrane fusion.
- Yin reports that previous attempts to determine the prefusion F protein structure were unsuccessful because the secreted protein without its transmembrane domain spontaneously folded to its postfusion state. To combat this, the authors appended an engineered GCN trimerization domain to mimic the transmembrane domain of the protein. The authors explain that this stabilized the trimer and reduced its fusogenicity. They also note that conceptually related constructs had been reported for the HIV Env and influenza virus hemagglutinin proteins. In the methods section of the paper, the authors also note that the furin cleavage site of the protein had been mutated to prevent intracellular processing.
- Figure 1 shows the structure of the PIV5 F protein with appended GCN trimerization domain in the prefusion conformation. Figure 2 shows the prefusion PIV5 protein structure and postfusion PIV3 protein structure side-by-side for comparison (see below). The diagrams reveal that the prefusion and postfusion conformation are strikingly different. The precise conformational changes are described under the heading "Comparison of the SV5 and hPIV3 F structures" at the bottom right of page 39 of the paper.

- In the same section of the paper on page 40, the authors note that "potentially related form of the RSV F protein have been observed in electron micrographs". Based on the prefusion and postfusion F structures, the authors propose a model of induction of membrane fusion by paramyxoviruses. This is set out and described in Figure 5.
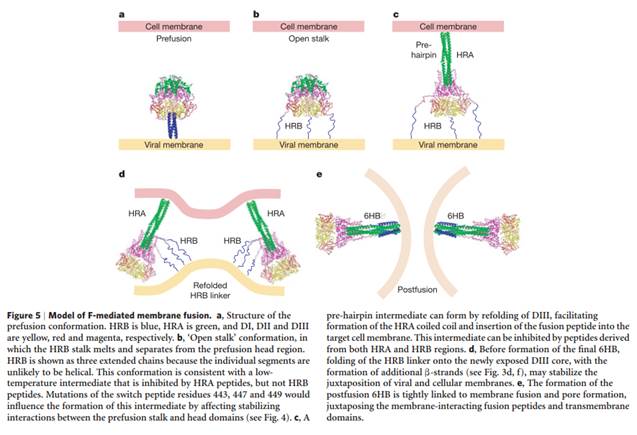
- The conclusions start on page 41 and a model for how fusion may occur is proposed (page 42, Figure 5). On page 42 in the final paragraph of the conclusions (which spans from the bottom of the left column to the end of the right column) the authors state that the folding of metastable proteins, such as the F protein, is not well understood and stabilising prefusion F using GCNt "may be important for the elucidation of other viral fusion protein mechanisms".
- Following the conclusion there is a methods section, which starts on page 42. The first paragraph "F protein expression and purification" discusses that the PIV5 construct they made with the GCNt trimerisation domain also had "the furin cleavage site ... mutated to prevent intracellular processing".
- This is an abstract from the 26th Meeting of the Annual Society for Virology that took place at Oregon State University in July 2007. The abstract reports on the generation of soluble RSV F glycoproteins. The authors are Supranee Chaiwatpongsakorn and Mark E. Peeples.
- The abstract begins by providing some background information on the RSV F and G proteins. It explains that the RSV F protein is the only viral protein required for infectivity and that as the RSV G protein is not required for membrane fusion, it is likely that the F protein has the ability to attach to target cells where it is triggered to initiate fusion.
- The abstract then refers to "recent structural studies" and explains that these have demonstrated that the paramyxoviral F protein undergoes a dramatic conformational change from the pre-triggered (virion) form to the post-triggered form to cause fusion.
- The authors note that they had computer modelled the prefusion and postfusion structures of RSV F and used those to suggest candidate triggering domains. They then describe the production of prefusion and postfusion soluble RSV F proteins and explain that the "the pretriggered form was fused to a trimerization domain to clamp its C-terminus and prevent premature triggering". The abstract reports that the proteins were secreted in a fully cleaved state. It reports that sucrose gradient tests were carried out and that incubation at 50°C caused the increased sedimentation of the prefusion form, suggesting it had undergone conformational change to the postfusion state. It further confirms that results from sucrose gradient testing indicate that both prefusion and postfusion forms were produced and that heat caused triggering.
- I approach my assessment of these allegations in four parts:
i) First, I must address some seemingly overarching arguments made by GSK.
ii) Second, I assess the allegations of obviousness over each piece of prior art in order to reach prima facie conclusions.
iii) Third, I address GSK's case on secondary evidence.
iv) Fourth, I state my overall conclusions.
- GSK argued that the obviousness analysis being advanced by Pfizer is the one which Technograph warns against in the following ways:
i) The Skilled Vaccinologist, at the priority date was faced with numerous potential approaches to vaccinating against RSV.
ii) Subunit vaccines had been researched over many years and, at least according to Dr Johnson, were not a favoured approach.
iii) There had been various manifestations of subunit F vaccines: subunit F vaccines on their own (now known to be postfusion), hybrid FG and shortly after the priority date hybrid F, G and M (matrix protein).
- In particular, GSK suggested that in order for the Skilled Vaccinologist to arrive at the invention, it was necessary that they:
i) Decide to revisit F subunit vaccines in preference to the more favoured approaches.
ii) Choose the preferred alternative approach to the subunit F vaccine is revisiting its structure rather than the manner of its presentation (adjuvants, modes of delivery etc).
iii) Recognise that the problem with existing F subunit vaccines, such as PFP, was that they were in the postfusion conformation, a problem not previously recognised in the literature.
iv) Consider that it might be possible to address this issue by making an alternative form of F subunit antigen, as opposed to just abandoning subunits in favour of live attenuated vaccines, vectored vaccines, or DNA vaccines.
v) Pursue an F subunit antigen in the prefusion form as opposed to the intermediate form or some other form.
vi) Find a source of the prefusion F protein.
- In common with their approach on other points, GSK's contentions I have just set out appear to be directed at a case of obviousness over the CGK. They do not grapple with the disclosure of each piece of prior art. I accept that there were many routes which had been and which continued to be researched by real-life teams in the RSV field, but GSK's points do not establish that the Skilled Team would have rejected the teaching of the prior art in this case. To the contrary, each piece of prior art would have provided the Skilled Team with sufficient motivation to follow its teaching.
- The final point is GSK's suggestion that the Skilled Team would not have been able to make a stabilised form of the prefusion F protein either at all or without undue burden, a point which I must return to later.
- Nonetheless I can address briefly the six points raised by GSK. Points i), ii), iv) and v) in [663] (as well as the three points in in [662]) are overcome, in my view, by what the Skilled Team does in the light of each piece of prior art and the CGK. My understanding is that each piece of prior art plus the CGK puts the Skilled Team in mind of an F subunit vaccine, and a necessary focus on structure to ensure stabilisation of the metastable prefusion form.
- GSK's third point is a central part of their case, but in my view it is a piece of advocacy and a clever attempt to reframe the debate in GSK's favour. There are three points:
i) First, this point oversimplifies the situation. It was not: postfusion bad, prefusion good. The literature suggests that postfusion subunit vaccines were still seen as viable for providing some protection, it's just that the prefusion form was hypothesised to provide better protection.
ii) The second point is that, in this case, the problem to be overcome was not recognition that existing F subunit vaccines were in the postfusion conformation. The problem was how to stabilise the prefusion form.
iii) The third point is that this proposition was never established in evidence, either by GSK's witnesses or by either Dr Johnson or Professor Weissenhorn agreeing that it was the problem in cross-examination.
- What remains are point vi) and GSK's point that the Skilled Team would not have been able to make a stabilised form of the prefusion F protein either at all or without undue burden. As I have indicated, I return to these later.
- This is the strongest documentary prior art. I have identified the skilled team above and resolved the issues over the CGK. I have addressed the disclosure of the Jardetzky Abstract and Slides above.
- To recap, the Jardetzky Abstract and Slides teach the use of a C-terminal trimerization domain to stabilize the F protein of the PIV F protein in its prefusion conformation.
- The difference between claims 1 and 5 of EP258 and the Jardetzky Abstract and Slides is the virus: RSV rather than PIV.
- The difference between claim 8 and the Jardetzky Abstract and Slides is the virus (RSV rather than PIV) and the use of the stabilisation RSV F protein antigen in the prevention or treatment of RSV-associated diseases.
- In light of my analysis of the CGK above, the skilled team would be aware of the analogy between RSV and PIV and would consider it obvious that they could also stabilize a prefusion F construct using a trimerization domain.
- They would have been confident that they could stabilize (as I have construed that term) a prefusion F construct using a trimerization domain.
- Even if they were not confident that it would be sufficiently stable to provide a vaccine candidate, they would have been confident that a construct could be stabilized in the sense of more stabilized than if it did not have a trimerization domain.
- The skilled team, reading the final point of the summary and conclusions slide, would be put in mind of a subunit vaccine for RSV F.
- On the assumption that the skilled team had made a prefusion F construct and had added a trimerization domain so that it was stabilized in the sense of being more stable than it would have been without that trimerization domain, if they had such a construct, it would be an obvious and routine matter to assess its immunogenicity using the techniques such as those described in the patent. The skilled team would be reasonably confident that such a construct would have some level of immunogenicity.
- The above applies to all of the relevant claims of EP710. Notably, the technical functional requirement of "stabilized" is only in claims 23 and 24. GSK's case appears to be that that requirement is inherently achieved by the modifications specified in claims 1, 10 and 22. The two structural features required by these claims are the use of a heterologous trimerization domain and the deletion of the furin cleavage sites.
- Pfizer's case is that removal or mutation of the furin cleavage sites would be an obvious thing to do to the skilled team. Prof Wilkinson agreed that the removal or deletion of such cleavage sites, if someone had identified where they are (and it is common ground that it was known where the furin cleavages sites were in RSV F at the priority date), then an obvious step would be to remove them in a hope to stabilise the polypeptide in which they are found, at the priority date.
- Dr Taylor's evidence was consistent with this - although she explained that she has read post priority papers on it so she does not know how obvious it would be. Once the skilled team had considered stabilizing the RSV protein with the trimerization domain, it would be an obvious step, if it was not stabilized enough or they wanted more stability, to take other stabilization steps. The deletion of furin cleavage sites was one such obvious step as Dr Taylor accepted.
- All the points I considered above apply with additional force in light of the oral disclosure in conjunction with the Slides. Even if, which I do not accept, Professor Jardetzky had unconsciously enhanced somewhat his account of what he said (which was GSK's criticism), the same conclusion would apply.
- I have identified the skilled team, the common general knowledge and the disclosure of Yin above.
- As to the inventive concept of EP258 it is to stabilise the prefusion conformation of the RSV F protein in a particular way, namely by use of a single polypeptide containing the F2 and F1 domains which is stabilised in the prefusion conformation by addition of a heterologous C terminal trimerization domain. The dispute on validity in terms of obviousness in relation to EP258 therefore boils down to:
i) whether or not it was obvious to add a trimerization domain to a recombinant RSV antigen, to 'stabilize' the prefusion conformation of the F protein and
ii) whether it would be obvious to use such an antigen as a vaccine.
- As for EP710, it is similarly:
i) whether or not it was obvious to add a trimerization domain and mutate/delete furin cleavage sites to 'stabilize' the prefusion conformation of the F protein and
ii) whether it was obvious to use such an antigen as a vaccine.
- As to Pozzoli question 3, the differences between Yin and the claims of the EP 258 Patent are the following:
i) The difference between claims 1, 5 and 6 of EP258 and Yin is the virus, which is RSV rather than PIV5 (both of which are paramyxoviruses).
ii) The difference between claim 8 of EP258 and Yin is also the virus (RSV rather than PIV5) and the use of the stabilized RSV F protein antigen in the prevention or treatment of RSV-associated diseases.
- The differences between Yin and the claims of the EP 710 Patent are the following:
i) The difference between claims 1, 10, 23 and 24 of EP 710 and Yin is the virus, which is RSV rather than PIV5 (both of which are paramyxoviruses).
ii) The difference between claim 22 of EP 710 and Yin is also the virus (RSV rather than PIV5) and the use of the stabilized RSV F protein antigen in the prevention or treatment of RSV-associated diseases.
- Turning to whether those differences constitute steps which would have been obvious to the person skilled in the art or do they require any degree of invention, I shall start by considering the skilled team's reaction to Yin, as that is important to have in mind when considering what (if anything) the skilled team would do in response.
- Dr Taylor's written position was to dismiss Yin on the basis that it is not specifically directed to RSV nor concerned with vaccine development, but this was based on her restricted view of the Skilled Team and their CGK.
- As a consequence of earlier work done on HIV and other envelope glycoproteins, the skilled team would be aware of and familiar with GCN4 and other trimerization domains as a matter of their CGK. In Yin the prefusion conformation of the PIV5 F protein (known to be analogous to that in RSV) was stabilized by using a C-terminal GCN4 trimerization domain. The skilled team would understand that Yin provides a rationale for why soluble proteins, which lacked the transmembrane and cytoplasmic regions would, when expressed, spontaneously fold or refold over time into the stable postfusion state.
- In oral evidence, Dr Taylor was prepared to accept that if you have a skilled team that was aware that the structure and conformation of PIV F was considered analogous to RSV F, then some of the teaching within Yin would have been considered transferrable to RSV.
- As a starting point many neutralizing epitopes were known to be conformational - Dr Taylor agreed with this point in cross-examination. The change in conformation would lead the skilled team to consider that it is likely that there would be a change in the epitopes or disruption to the conformational epitopes that might be recognised in the prefusion form - it is a possibility that there will be new epitopes exposed on the prefusion compared to the postfusion form.
- Dr Taylor further agreed that stabilisation of the prefusion conformation would also allow the differences between epitopes on the pre and postfusion form to be investigated. This is important for the earlier claims of both of the Patents, which do not claim the use of the construct as a vaccine.
- Ultimately, she accepted that if the prefusion form of RSV was seen as a target for vaccine design at the priority date, then she would accept it would be an obvious first step in the light of the teaching of Yin to seek to stabilize the prefusion form of RSV F with a C-terminal trimerization domain as Yin describes.
- As for the deletion of furin cleavage sites claimed by EP258, Dr Taylor agreed that if you were seeking to stabilize prefusion conformation of an F construct, or any further stabilisation, deletion of furin cleavage sites would be an obvious next step.
- I agree that the idea that a C-terminal trimerization domain could be used to stabilize the prefusion conformation of a paramyxovirus F glycoprotein had been demonstrated in Yin (and in any event was published in a standard textbook at the time, Fields).
- The ASV abstract informs the skilled team that the author has made prefusion and postfusion soluble RSV F protein.
- Dr Taylor noted that it teaches that the prefusion form was made by fusing with a C-terminal trimerization domain to prevent premature triggering. Again, unsurprisingly, she agreed that it would disclose to the skilled person the idea of using a soluble RSV F protein stabilized in the prefusion conformation by use of a C-terminal trimerization domain. The ASV Abstract does not say so in terms, but it is implicit that it has an F1 and F2 domain.
- An RSV F subunit would have been viewed as an obvious target for vaccine research. If that is correct, then the ASV abstract would provide a clear and obvious path to providing a stabilized RSV prefusion form of F, as Dr Taylor agreed.
- As for the additional structural feature of EP710, she agreed that if you stabilized in this way (i.e. with a trimerization domain) and it was not stabilized enough, she accepted that although it does not mention the deletion of furin cleavage sites, that would be an obvious next step to take. A skilled team considering a subunit F vaccine would look at the ASV abstract and think it is a good idea to try, based on that, to stabilize the RSV F in its prefusion conformation.
- Before I reach any conclusions on the allegations of obviousness over each piece of prior art, I must consider GSK's case on the secondary evidence.
- In Schlumberger Holdings Ltd v Electromagnetic Geoservices AS [2010] EWCA Civ 819; [2010] RPC 33, Jacob LJ considered the role of secondary evidence in some detail:
"[77] It generally only comes into play when one is considering the question 'if it was obvious, why was it not done before?' That question itself can have many answers showing it was nothing to do with the invention, for instance that the prior art said to make the invention obvious was only published shortly before the date of the patent, or that the practical implementation of the patent required other technical developments. But once all other reasons have been discounted and the problem is shown to have been long-standing and solved by the invention, secondary evidence can and often does, play an important role. If a useful development was, in hindsight, seemingly obvious for years and the apparently straightforward technical step from the prior art simply was not taken, then there is likely to have been an invention."
- Whilst Pfizer pointed to a number of cases where there are general statements in the authorities noting that secondary evidence is, indeed, secondary to primary evidence, it is worth bearing in mind Jacob LJ in Schlumberger at [85]:
"[85] It would be wrong to read this decision as saying that secondary evidence is always of minor importance. That would be to throw away a vast mass of jurisprudence, including many House of Lords cases, (e.g. Vickers, Sons & Co v Siddell and Technograph). It would indeed involve disregarding some of the approach actually used in Mölnlycke."
- I have found valuable the following observations by Meade J in Gilead v NuCana [2023] EWHC 611 (Pat), where he applied the distinction between primary and secondary evidence to an issue of undue burden:
'442. Where the issue is one of obviousness the courts in this jurisdiction are very used to separating primary evidence (that of the experts) from secondary evidence, such as the experience of real world workers who did or did not make the invention, and applying appropriate caution to the latter, based for example on whether they represented the ordinary skilled person, whether they had the cited art, and so on.'
'446. I also identify at this stage that the usefulness of secondary evidence must depend in significant part on how complete and how testable it is. In the present case I did not hear oral evidence from any of the real world workers relied on, and the documentary record is patchy, including because the documentation created at the time was poor (in the case of Mr Clark). This makes it especially hard to assess why the workers in question succeeded or failed, as the case may be. Secondary evidence on obviousness is often discounted by a trial judge on the basis that it is simply unknown why (for example) the invention was not made before and in my view the same should apply to undue burden, as part of the overall exercise of assessing the secondary evidence.'
- The notion that the usefulness of secondary evidence must depend on how complete and how testable it is brings to mind the analysis of Birss J. in Accord v medac [2016] EWHC 24 (Pat) where he introduced the point as follows:
65. Finally I will mention secondary evidence. An important part of medac's argument was to ask the rhetorical question - if it was obvious, why was it not done before? It was common ground that while this is a form of secondary evidence, as compared to the primary evidence being the reasons given by the experts for their opinions, such secondary evidence has its place and in a proper case can be powerful.
- Having considered medac's submissions on the secondary evidence in that case, Birss J. concluded they were legitimate points which carry weight overall. He nonetheless concluded the invention was obvious over a piece of prior art called Russo. The secondary evidence arguments 'are not strong enough to overcome what is a powerful obviousness case...'
- The patent in issue was for the use of a formulation containing methotrexate in a concentration of about 50mg/ml, when known concentrations were at a lower level (no more than 25mg/ml) - these had been available since 1995. Russo was published in 2000, yet the invention was not made until medac did the work leading up to the Patent which was filed in 2006.
- Although it is clear that the secondary evidence in that case was not complete, Birss J. analysed the position in Germany and the UK. The factors which appeared to be influential in Germany were that (i) the art was just more focussed on new biologic agents (ii) methotrexate was a generic medicine so margins were tight, so although the commercial factors were not addressed in the evidence, Birss J. was inclined to conclude that it only became commercially attractive to go to the trouble of producing a 50mg/ml product towards the priority date, after once subcutaneous administration became very well established on a large scale. In the UK, clinicians did not encounter the problem of pain caused by large subcutaneous injections because they were not the ones administering the drug, and there was no evidence from nurses who did.
- As for Russo, although it appeared in a well-read journal, it was not CGK, neither expert recalled reading it and no evidence was called from anyone who read it at the time.
- Overall, that analysis is a perfect illustration that the consideration of primary and secondary evidence requires a highly nuanced approach which is only possible where the Court has confidence that it has received sufficiently complete evidence of what real-world teams were doing.
- In the 'normal' case (to the extent that any Patent case is normal), the Court has evidence of the real-world teams working on the problem in question and, assuming they were aware of the prior art relied upon, failing to come up with the invention before the priority date.
- This case is somewhat different. In this case the Jardetzky art was made available to the public in (late) October 2007, about 8 weeks or so before the priority date. The ASV Abstract was published in July 2007. The earliest publication in time is Yin (January 2006), almost two years before the priority date. Yin (or at least parts of it) were agreed to be CGK, so the secondary evidence arguments have the most force in relation to Yin.
- So, although I can proceed on the basis that Yin was known to the art and there was plenty of evidence of the other approaches being taken, GSK did not lead any evidence that there were any real-world teams who decided to pursue a sub-unit vaccine approach. Instead, the unstated assumption underlying GSK's case was that there were real-world teams trying and failing. I must assess whether this assumption is correct.
- GSK in their closing skeleton argument introduced for the first time, their full case on secondary evidence, devoting no less than 26 pages to this topic. Despite not being pleaded, it clearly formed a key part of their case.
- GSK submitted that it is highly persuasive that there is no reference to the problem (that existing subunit vaccines were in the postfusion form - which I will refer to as 'the stated problem') or to the solution (stabilise the prefusion form) in the large number of papers and reviews available to the court; not only before the priority date but between the priority date and 2013, when McClellan 2013B was published in Science
- Throughout the relevant background was that there was an active RSV community, a motivation to produce alternative and improved vaccines and a long felt want in that regard. The primary prior art was generally known in the sense that Professor Jardetzky gave his presentation to the 'great and the good' of the RSV field, and Pfizer said that Yin was CGK. GSK did not accept that Yin was CGK to the vaccinologist but accepted that the broad concepts as reported in Fields were known.
- Furthermore, GSK relied on the fact that the prefusion and postfusion forms of RSV were known from 2000. They accepted that more detail was provided in Yin, but the basic concept of a major refolding of the F antigen on activation was known in the art and described in Cane.
- It is convenient to address GSK's case and the materials they relied upon in two parts: pre-priority and post-priority. Although the cross-examination was conducted by reference to a large number of papers, in their closing, GSK were selective as to which ones they chose to rely upon.
- GSK's argument started with references to the textbooks.
- First, the discussion in Cane as to the strategies which had been and were being pursued. As GSK pointed out, the eminent authors discuss F subunit vaccines at length, discuss strategies that are being investigated to improve them and yet do not suggest there might be a structural problem with the PFP or other subunit forms of F antigens.
- Second, Chapters 41 (on Paramyxoviridae) and 46 (on RSV) of Fields. As I pointed out above, Yin (and Yin 2005) feature extensively in Chapter 41. Chapter 46 contains a discussion of subunit vaccines at p1635, with a section on Perspectives and concluding with 'Many important and fundamental questions'. Again, as GSK submitted, the authors (Collins & Crow) fail here or elsewhere to recognise the problem with F subunit vaccines is that they are in the postfusion form. Again, they suggest the way forward is live attenuated viruses.
- Counsel also referred to a number of review articles which surveyed the whole field of published RSV research. Apart from indicating there were a large number of possible routes being explored to either vaccinate or ameliorate the effects of RSV, these review articles were not particularly illuminating and did not add to the picture painted in Fields which was the most up to date publication at the priority date.
- I can start with Fields (6th Edition, 2013). GSK anticipated that Pfizer might argue that what was obvious had not made it into the textbooks published before the priority date, so they cross-examined Dr Johnson on the next edition of Fields, published in 2013 - some 5 years after the priority date. The RSV and metapneumovirus chapter was written by Peter Collins and Ruth Karron, whom Dr Johnson agreed were 'super-skilled' and at the 'cutting edge' on the subject matter. Naturally it contains a discussion of the main classes of vaccines. In relation to subunit vaccines, the authors describe PFP and BBG2Na before stating this:
"Increasing knowledge of the structure and antigenic properties of the HRSV F protein and improved methods of expression and purification may allow for the production of more stable and more immunogenic HRSV F vaccine preparations. As of this writing, the only HRSV subunit vaccine currently being evaluated in clinical trials is an F protein particle vaccine developed by Novavax (clinicaltrials.gov NCT01290419), which is currently undergoing phase 1 evaluation in healthy adults. In other recent work, the HRSV F protein was engineered to remove the fusion peptide, transmembrane region, and cytoplasmic tail, yielding an expressed protein that formed a postfusion trimeric structure that was homogenous, stable, and highly immunogenic.378, 536" (emphasis added)
- Those references were 378 - McLellan 2011 and 536 - Swanson 2011.
- Dr Johnson agreed that this would have been a 'perfect moment' for the authors to suggest stabilising in the prefusion form, but that idea was missed.
- This Chapter in Fields 2013 concludes with a forward-looking section entitled "Perspectives", where the authors speculate on possible ways forward for RSV vaccine design. They do not suggest a prefusion form, as Dr Johnson agreed. GSK submitted that the problem had not been identified by these authors by the time this chapter Fields 2013 was written: there was no appreciation that the failure of the RSV F subunit vaccines resulted from their postfusion conformation. Indeed, Fields 2013 positively advocated a vaccination approach with the postfusion form.
- GSK also relied on further review papers published after the priority date which, they said, still failed to identify the starting point needed to look towards the invention. By this they meant a continuing failure to appreciate the stated problem. I refer to this as 'the stated problem' because it is necessary to assess whether this was the problem standing in the way of the development of a RSV F subunit vaccine stabilised in the prefusion conformation.
- In order to make this assessment, I found it necessary to review more than just the papers which GSK selected to rely upon. Generally, GSK were correct that the review articles did not identify the stated problem. However, as regards the papers reporting research progress, it is clear there was interplay between several different groups. In this regard, GSK emphasised that the research groups were 'super-skilled' and I take account of that point. However, when it comes to secondary evidence, it is not restricted to a level equivalent to CGK at the priority date.
- First, a 2007 review by Meyer entitled Human and bovine respiratory syncytial virus vaccine research and development. Subunit vaccines are discussed from pp11-15. Under 'Conclusions', the authors take a prospective view on vaccine development and, as GSK submitted, they do not even begin down the pathway of considering the antigenic significance of the two conformations.
- Second, Murata 2009 reviews the avenues of vaccine research, including PFP, BBG2Na and an F/G/M vaccine from the subunit research. Again, having surveyed the existing F subunit vaccines, it fails to make any hints in the direction of the conformation of the F protein (still less identifying the postfusion form as the origin of their failure).
- McLellan 2010A (December 2010) is entitled 'Structure of a Major Antigenic Site on the Respiratory Syncytial Virus Fusion Glycoprotein in Complex with Neutralizing Antibody 101F'. As indicated in the abstract, the team investigated the mechanism of antibody-mediated RSV neutralization by the monoclonal antibody 101F which binds a linear epitope in the RSV fusion glycoprotein.
- Antigenic sites II and IV were known from the Calder paper and to exist in both the prefusion and postfusion conformations, and it was also known that site IV was the target for 101F. As stated and illustrated in Fig 3, '101F is predicted to bind with similar affinity to both the pre- and the postfusion conformations'.
- As the authors stated in the Introduction:
'We undertook structural and functional studies of the inter- action between 101F and its epitope on the RSV F glycopro- tein to investigate the mechanism of antibody-mediated RSV neutralization. Here we present the crystal structure of the antigen-binding fragment (Fab) of 101F in complex with its F glycoprotein-derived epitope peptide. The structure defined the length of the linear epitope and allowed for modeling of 101F binding to pre- and postfusion F trimers. Hypotheses based on these models were tested to investigate the mechanism of 101F neutralization and the extent of the epitope. These results are analyzed and discussed in the context of known antibody escape mutations, mechanisms of antibody-mediated virus neutralization, and applicability to epitope-specific vaccine design.'
- In the Discussion section, the authors demonstrate they were well aware of (a) the possibility of neutralizing antibodies blocking various steps in the virus entry process but also (b) that 101F does not prevent triggering:
'During the entry process, there are several steps which neutralizing antibodies can block, including attachment, triggering, and transition of the fusion glycoprotein to the postfusion state. We have demonstrated that 101F does not block virus attachment over a wide range of concentrations (Fig. 5B), which agrees with recently published data showing a similar result at a single antibody concentration (29). We have also shown that 101F is capable of preventing infection once the virus has attached to the cell (Fig. 5C). This narrows the window of 101F neutralization to some point between triggering of the fusion glycoprotein and transition to the postfusion state. In addition, since our modeling studies show that 101F binding is compatible with both the pre- and postfusion states (Fig. 3), 101F is predicted to bind all forms of the fusion glycoprotein, including intermediates. Thus, we propose that 101F also does not prevent triggering but rather prevents adoption of the postfusion conformation due to its bulk in the context of the cell and viral membranes.'
- GSK pointed out that Yin (ref 46) is referred to in this passage in the introduction, just before the passage cited above:
'This epitope [the context is antigenic site IV] is C-terminal to the cysteine-rich region and is part of domain II, which in homologous paramyxovirus F glycoproteins remains structurally unchanged between pre- and postfusion conformations (46).'
- In cross-examination, Dr Johnson agreed that the authors had identified that 101F binds to both the pre- and postfusion forms and so were contemplating antibodies which neutralised the fusion process by binding to anything between attachment and fusion. As I have already stated, that was known from the Calder paper. On that basis, GSK submitted that 'the first turn which these authors take based on their consideration of Yin 2006 is in the direction of indifference as between the pre, post or intermediate conformations.' I do not agree that this is the only conclusion that can be drawn. In this paper, the authors appear to have been following up on some earlier research from the Melero group relating to 101F, reported in Wu (ref 44) 'Characterisation of the epitope for anti-human respiratory syncytial virus F protein monoclonal antibody 101F using synthetic peptides and genetic approaches'.
- McLellan 2010B is entitled 'Structural basis of respiratory syncytial virus neutralization by motavizumab' published in February 2010. This was a paper which Pfizer put to Dr Taylor in cross-examination. She agreed that the authors were investigating differences between palivizumab and motavizumab and to do that they wanted to compare the binding of those two antibodies to RSV.
- To vizualise the binding of motavizumab to full-length F glycoprotein, a model was generated based on the structure of prefusion PIV5, referring to Yin. Experimentally, they expressed and purified a soluble form of RSV F in a form similar to PIV5 F used in the modelling. The furin cleavage sites were mutated and a fibritin trimerization domain was appended to the truncated C terminus to keep the protein in a trimeric, pre-fusion conformation. This stabilized RSV F glycoprotein was referred to in the paper as RSV F0 FD.
- Dr Taylor agreed that when McLellan wanted to stabilize RSV F in its prefusion conformation, at this point for investigating different bindings of antibodies, before investigating the postfusion conformation he immediately took the teaching of Yin and recreated it in RSV, taking two steps: one adding a fibritin trimerization domain (a foldon domain) and two, deleting the furin cleavage sites
- In closing, GSK sought to dismiss the relevance of this paper, suggesting (a) that the reason the authors prepared a stabilised prefusion form of the RSV F protein was to confirm that motavizumab did not bind to it and (b) that this was all of a piece with their other contemporaneous publications which described the intermediate forms and positively suggest a vaccination approach favouring the postfusion form.
- I do not agree this is the only conclusion which can be drawn. This paper does raise a number of questions: first, why, having apparently achieved a stabilised form of prefusion RSV F, McLellan's group did not immediately investigate its potential as a vaccine; second, why it seems to have taken another 2 years to achieve that; and third, why McLellan's group seem to have continued to investigate the binding of antibodies to RSV. It may have been the case that they had particular reasons for those investigations. The answers to these questions could have been explored if someone from the McLellan group had given evidence.
- This paper is inconsistent with GSK's suggestion that McLellan's first reaction to Yin was to go for a postfusion conformation. Instead, it seems to be neutral. Dr Taylor agreed that if there was motivation to investigate the prefusion conformation, it would have been obvious in the light of Yin to stabilize it as Yin did, and indeed to add a fibritin trimerization domain.
- Swanson 2011 is entitled 'Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers'. Swanson and his colleagues are identified as from Novartis.
- Swanson 2011 starts by explaining that F is a promising antigen for RSV candidate vaccines and that the basic features of RSV F are shared with the fusion glycoproteins of other members of the Paramyxoviridae, such as PIV3, PIV5 and NDV. It then describes the process of activation of RSV F for membrane fusion and continues:
'The prefusion and postfusion forms of RSV F each have potential shortcomings as vaccine antigens. Large structural differences between the lollipop-shaped prefusion F trimer and the crutch-shaped postfusion F trimer are apparent even at the resolution of electron microscopy of negatively stained specimens, suggesting that prefusion and postfusion F may be antigenically distinct (11). To prevent viral entry, F-specific neutralizing antibodies presumably must bind the prefusion conformation of F on the virion, before the viral envelope fuses with a cellular membrane. Therefore, it might be expected that RSV F must be presented in the prefusion conformation to elicit neutralizing antibodies efficiently. However, prefusion F is a "metastable" structure that readily rearranges into the lower energy postfusion state, which aggregates due to exposure of a hydrophobic fusion peptide (12), and efforts to generate a soluble, stabilized prefusion F subunit antigen have not yet yielded candidates suitable for testing in humans.'
- Having made those observations, the main point of the paper is as summarised in the abstract that:
'We have generated a homogeneous, stable, and reproducible postfusion RSV F immunogen that elicits high titers of neutralizing antibodies in immunized animals. The 3.2-Å X-ray crystal structure of this substantially complete RSV F reveals important differences from homology-based structural models. Specifically, the RSV F crystal structure demonstrates the exposure of key neutralizing antibody binding sites on the surface of the postfusion RSV F trimer. This unanticipated structural feature explains the engineered RSV F antigen's efficiency as an immunogen. This work illustrates how structural-based antigen design can guide the rational optimization of candidate vaccine antigens.'
- In their closing submissions on secondary evidence, GSK chose to mention Swanson 2011 only by reference to the one sentence summary of it in Hurwitz 2011 (see below). However, on a slightly closer analysis, it appears that the Novartis group were continuing their efforts to generate a soluble, stabilized prefusion F subunit antigen. It would appear the problem lay in achieving stabilization of the prefusion form.
- McLellan 2011 (August 2011) is entitled 'Structure of Respiratory Syncytial Virus Fusion Glycoprotein in the Postfusion Conformation Reveals Preservation of Neutralizing Epitopes'. It appears to be a continuation of their research into neutralizing monoclonal antibodies (in this instance, palivizumab, motavizimab and 101F) targeting antigenic sites II and IV. As stated in the abstract: 'The structures of these sites as peptide complexes with motavizumab and 101F have been previously determined, but a structure for the trimeric RSV F glycoprotein ectodomain has remained elusive. To address this issue, we undertook structural and biophysical studies on stable ectodomain constructs.'
- GSK drew attention to this passage in the Discussion (in which ref 44 is Yin):
"Thus, 101F and motavizumab can likely bind the fusion glycoprotein in the prefusion, postfusion, and intermediate states, which results from their epitopes residing in domains that are not expected to undergo large structural rearrangements during the fusion process (44)."
- GSK again submitted that 'Their interpretation of the crystal structures presented in that paper is in favour of an indifference between the different conformations'.
- Hurwitz 2011, entitled 'Respiratory syncytial virus vaccine development'. This is an extensive review of the whole RSV field, with 18 pages of text and citing 261 papers. Under the heading 'Purified F protein', the final sentence says 'Purified F proteins remain a topic of interest, with recent attention paid to the protein's postfusion structure [169].', where reference [169] is to Swanson 2011. GSK submitted as follows:
'It is all of a piece with the abovementioned papers, showing that an appreciation of the problem lying in the postfusion conformation of the existing F subunit vaccines formed no part of the prevailing view. Additionally, we remark that the fact that the report of postfusion was picked up so quickly in the review papers further tells against the suggestion that an approach to vaccination based on conformation had been taken earlier.'
- Schmidt 2011, entitled 'Progress in Respiratory Virus Vaccine Development' is a general review article which covers the full breadth of vaccination avenues. It records the Sanofi Pasteur F, G, M subunit vaccine in vaccine trials. GSK submitted that nowhere does it suggest seeking to vaccinate with the prefusion form of RSV F. Its conclusion states: "No one can tell whether live attenuated, subunit, VLP, replicating, or nonreplicating vectored vaccines will turn out to be successful in phase 3, but hopefully this new competition will help to bring much needed respiratory vaccines to the market sooner. Every year without them will be a lost year, especially for those who don't receive the care they need." Dr Johnson agreed that Schmidt (2011) is a fairly thorough review which gives a feel for all the different routes being pursued in the hope of getting an RSV vaccine. GSK also submitted that its review gives the impression that the authors were not particularly attracted to the subunit approach in view of the failures of the PFP, BBG2Na and other existing F subunits.
- Collins and Melero (2011), by authors who are acknowledged leaders in the field. This paper, published in December 2011, contains a broad, 48-page review of RSV entitled "Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years". GSK submitted the title rather gives it away: things are still crazy as the field has not alighted on a suitable vaccine. From pages 18-21, the authors cover each of the major vaccines under consideration. In the passage bridging pages 20-21, the authors state
"More recently, as already noted, a post-fusion form of the F protein was produced with deletion of the major hydrophobic regions (McLellan et al., 2011; Swanson et al., 2011). Importantly, this expressed protein forms stable trimers that were recognized by a number of neutralizing MAbs. In rodents, this antigen induced high titers of neutralizing serum antibodies and protection against RSV challenge. This may represent an improved RSV subunit vaccine." (emphasis added)
- Perhaps not surprisingly in view of the common co-authorship of Peter Collins, this passage is to the same effect as the last sentence from Fields 2013.
- McLellan 2013A is entitled 'Structure of RSV Fusion Glycoprotein Trimer Bound to a Prefusion-Specific Neutralizing Antibody' and was published in Science on 31 May 2013. The abstract provides a good summary of the content of the paper:
'The prefusion state of respiratory syncytial virus (RSV) fusion (F) glycoprotein is the target of most RSV-neutralizing activity in human sera, but its metastability has hindered characterization. To overcome this obstacle, we identified prefusion-specific antibodies that were substantially more potent than the prophylactic antibody palivizumab. The cocrystal structure for one of these antibodies, D25, in complex with the F glycoprotein revealed D25 to lock F in its prefusion state by binding to a quaternary epitope at the trimer apex. Electron microscopy showed that two other antibodies, AM22 and 5C4, also bound to the newly identified site of vulnerability, which we named antigenic site Ø. These studies should enable design of improved vaccine antigens and define new targets for passive prevention of RSV-induced disease.'
- McLellan 2013B is entitled 'Structure-Based Design of a Fusion Glycoprotein Vaccine for Respiratory Syncytial Virus and was published in Science on 1 November 2013. The approach they took to achieve a stabilised form of the prefusion F is described in the following passage, to which I have added the key papers which are referenced:
'The proven success of palivizumab (3) has spurred vaccine efforts aimed at eliciting protective RSV F–directed antibodies. These efforts have been complicated by the conformational diversity of RSV F (4–8 [including McLellan 2011, Swanson 2011 and McLellan 2013A]), a type I fusion glycoprotein that merges virus and host-cell membranes by using the difference in folding energy between two substantially different states: a metastable state adopted before virus-cell interaction (prefusion) and a stable state that occurs after merging of virus and cell membranes (postfusion). Both states exhibit epitopes targeted by neutralizing antibodies, and postfusion RSVF is being developed as a vaccine candidate (6 [Swanson 2011], 9). Recently, however, the major target of RSV-neutralizing antibodies elicited by natural infection was found to reside primarily on the prefusion conformation of RSV F (10 [Magro 2012]). Antibodies such as 5C4 (7 [McLellan 2013A]), AM22, and D25 (11, 12) are substantially more potent than palivizumab and target antigenic site 0/ (zero), a metastable site located at the membrane-distal apex of the prefusion RSV F trimer (7 [McLellan 20132A]).
To enhance elicitation of similarly potent antibodies, we engineered soluble variants of RSV F with stably exposed antigenic site 0/. These variants were characterized antigenically and crystallographically and tested for immunogenicity in mice and nonhuman primates (rhesus macaques).
Structure-Based Vaccine Strategy
We and others have engineered antigenicity (13–17) through structure-based design of the epitopes recognized by template neutralizing antibodies. For example, the crystal structure of motavizumab (a variant of palivizumab) bound to its F glycoprotein epitope (18 [McLellan 2010]) allowed us to create epitope scaffolds, which stably presented the motavizumab epitope on heterologous proteins (19 [McLellan 2011]). Although motavizumab-epitope scaffolds could elicit immune responses that recognized F, substantial neutralizing activity was not induced (19). We hypothesized that instead of a single epitope recognized by a single template antibody, it would be advantageous to present a "supersite" (20), comprising a collection of overlapping epitopes recognized by multiple antibodies. Even more preferable would be for such a site to be ultrasensitive to neutralization. These considerations led to a "neutralization-sensitive site" strategy: (i) to identify a viral site targeted by multiple antibodies with extremely potent neutralizing activity, (ii) to determine the structure of the site in complex with a representative antibody, (iii) to engineer the stable presentation of the site in the absence of recognizing antibody, and (iv) to elicit high-titer protective responses through immunization with engineered antigens that stably present the neutralization-sensitive site (fig. S1).
Engineering of RSV F Antigens
Antigenic site Ø was chosen as the target site because of its recognition by RSV-neutralizing antibodies that are 10- to 100-fold more potent than palivizumab (7, 11, 12). We previously determined the structure of antigenic site Ø in complex with the D25 antibody (7). Structure determination involved appending the T4-phage fibritin trimerization domain ("foldon") (21, 22) to the C terminus of the RSV F ectodomain (5) and binding of the prefusion-specific D25 antibody. Although these approaches stabilized antigenic site Ø, D25 binding sterically occluded the target site. To stably present antigenic site Ø in the absence of D25, we retained the C-terminal trimerization domain and combined it with other means of stabilization, including the introduction of cysteine pairs or cavity-filling hydrophobic substitutions.'
- The final article referred to was entitled 'RSV Vaccines that work?' published in The Scientist Magazine in 2023. This appears to have been written on the basis of an interview with Barney Graham (and others) and it recounts the progress made towards a total of 6 Phase 3 Clinical Trials being undertaken in the US - 5 using prefusion F and the sixth using prefusion F with postfusion F and other proteins. The article also reports on 17 more Phase 1 and 2 vaccine trials targeting a variety of RSV proteins. It also reports on the effectiveness of both the GSK and Pfizer vaccines.
- The article contains a potted history of the development of research into RSV, starting with its isolation from chimpanzees in 1956 and then relating the disastrous foray into FI-RSV. A clinician is reported as saying 'for a long time people didn't dare to develop RSV vaccines', but the article reports that by the 1990s and 2000s some researchers had returned to working on RSV vaccines, 'typically using the F protein in its so-called post-fusion conformation'. This article provides the most detail about the route taken by McLellan. It continues:
'In 2012, however, a group in Spain showed that the majority of neutralizing activity in rabbits inoculated with a recombinant vaccinia expressing the F protein targeted the flighty pre-fusion form of F, not the stable post-fusion form. Not much was known about pre-fusion F because it was so transient, but soon afterwards, Graham and McLellan and their teams at NIAID's Vaccine Research Center figured out the structure of the pre-fusion F protein as it was bound to a powerful antibody called D25. In particular, Graham and colleagues saw that D25 and other strong neutralizing antibodies attach to the apex of the pre-fusion F protein, each at a different angle, interfering with the protein rearrangement required for the virus to fuse with and enter cells. They named this apical region antigenic site 0, and obtained high-resolution X-ray diffraction data on the complex's crystals, solving the structure with molecular replacement.
Because of its location on the pre-fusion F's apex, antigenic site 0 is accessible to antibodies even on the crowded surface of a virus, helping to explain why the strongest natural antibodies to RSV target pre-fusion F. "That's when we really got serious about trying to do the protein engineering steps to stabilize the molecule in the pre-fusion form," in order to use it as a vaccine antigen, Graham says.
Within a year, they'd cracked it. In a 2013 Science paper, the researchers reported that if they added cysteine residues to certain sites and filled some cavities in the protein structure, the protein remained in the pre-fusion state. Injecting this stabilized pre-fusion F protein, which they called DS-Cav1, into mice and macaques generated an RSV-specific neutralizing antibody response many times higher than what is needed to thwart RSV infection. "It was much more immunogenic in terms of inducing neutralizing activity than anything we had [ever seen] before," says Graham, who is named with McLellan, Kwong and others as a coinventor on patents for pre-fusion F protein antigen design, and consults for RSV vaccine developers.'
The words underlined above were hyperlinks: the first was to Magro 2012 and the second to McLellan 2013A.
- Although I have not benefitted from evidence from anyone in the McLellan group, the available information from the various papers I have reviewed indicates that the key difficulty which the McLellan group had to overcome was to stabilize the prefusion F protein, but they were only able to achieve this after they had identified the important antigenic site 0. This resulted from work reported in Magro 2012 to the effect that 'the major target of RSV-neutralizing antibodies elicited by natural infection was found to reside primarily on the prefusion conformation of RSV F'.
- Swanson 2011 also indicates the key difficulty which the Novartis group had to overcome was stabilisation (and see further below).
- These points, together with the teaching in the Patents and the approach taken to stabilize Pfizer's RSV PreF, indicates that there seem to be numerous ways to stabilize the prefusion F protein and different ways to approach the problem. For example, it appears that the route taken by McLellan depended on the identification of the new antigenic site 0. It also suggests that stabilizing the prefusion F protein is not as simple as merely attaching a GCN4 trimerization domain to the C-terminal of the F1 domain (cf EP258). In other words, it seems likely that the degree of stabilization afforded by merely attaching a GCN4 trimerization domain to a prefusion F construct does not provide a sufficient degree of stabilization of the prefusion F protein for practical use as an immunogen or as a vaccine, even though it might provide some degree of protection.
- The implicit assumption in GSK's case on secondary evidence was that the published literature provides an accurate picture of the reaction in the art to Yin and the Jardetzky disclosures. However, it is necessary to take into account a number of features of the situation.
- First, I have not had the benefit of anyone from either the McLellan or Melero groups explaining their work, how much time they were able to devote to RSV research (bearing in mind that I understood Jason McLellan's primary role to be in HIV research), their research interests in RSV or, for that matter, their reaction to the prior art pleaded in this case.
- Second, GSK's case on secondary evidence was never pleaded which has had at least three important consequences:
i) First, the case put in closing submissions appears to me to rely on a different selection of papers to those which were raised in Dr Johnson's cross-examination. In other words, the case was able to change without the constraint which would ordinarily be provided by it having to have been pleaded.
ii) Second, the lack of a pleaded case largely deprived Pfizer of a proper opportunity to address it (let alone investigate what evidence they might have been able to lead in response to it) although Pfizer were able to react to this case to some degree.
iii) Third, as I have already indicated, it is necessary to pay very careful attention to the few pieces of positive evidence which Dr Johnson was able to volunteer in the course of the cross-examination which was, as one might expect, closely focussed on what the cross-examiner was seeking to establish.
- Third, and in that vein, one of the most revealing answers in Dr Johnson's cross-examination was her response to Counsel's suggestion that 'the penny did not drop until 2013'. Her response was 'It did not produce any data until 2013'. The import of that answer cannot, in my view, be underestimated because it serves as a reminder that the published literature cannot reflect everything that is going on in the art. There are two immediate examples which can be cited:
i) First, my attention was not drawn to any publications emanating from GSK or the inventors of the Patents.
ii) My second example concerns the work of the Novartis group, in which Swanson and Dormitzer appear to have been the principal participants. Swanson 2011 was published in June 2011, having been submitted in April 2011. As I mentioned above, in that paper the authors reported on a postfusion antigen but nonetheless indicated they still considered that prefusion would be a better way to go ('to elicit neutralizing antibodies efficiently'). Yet, it appears that Novartis had already filed priority documents claiming a prefusion antigen. Pfizer drew attention to P1EP490 and P2EP490, with the named inventors as Kurt Swanson and Philip Dormitzer. P1EP490 was filed on 15 July 2009 and P2EP490 was filed on 12 January 2010 by Novartis. Novartis therefore appeared to have had a team achieving results on prefusion F in 2009 which indicates they had been working at that well before 2009. My attention was not drawn to any paper in which the Novartis group published the results of their success in stabilising F in the prefusion form and creating a viable vaccine.
- I draw attention also to the fact mentioned in the 2023 article that Jason McLellan was a co-inventor of patents for pre-fusion F protein antigen design. Although this is speculation, it raises the possibility that his publication of work on the prefusion form may have been affected by the requirements to get patent protection in place. Without evidence from someone in the McLellan group, we do not know.
- Dr Johnson was asked about the length of time it would take to see mention of work being done on a prefusion subunit vaccine. The question was posed on the basis of someone in the art who had (a) an interest in subunit vaccines and (b) attended Professor Jardetzky's presentation. Dr Johnson responded by saying a minimum of three years, unless you had a major lab that had the skilled team in place (i.e. with structural biology expertise). So, she envisaged the first signs emerging in about 2010, but only if the researchers chose to publish.
- Counsel responded to this by taking Dr Johnson to McLellan 2010A, and his point was that in that paper the authors did not propose stabilising in the prefusion form.
- The riposte to that was that prior to 2010, Jason McLellan was working in HIV. He joined Barney Graham's group at the NIH in 2010 but space was limited in the HIV lab so he was given a seat in the RSV lab. This indicates that Jason McLellan only really took an interest in RSV in 2010. On that basis, the success published in McLellan 2013B conforms to Dr Johnson's prediction.
- Fourth, it is apparent that the discussions in the literature take place at broadly two different levels. The numerous review articles provide, in the main, a very brief summary of each paper in the field - often a single sentence - because they have a lot to cover. The textbooks also have a lot to cover, but the extensive reference to Yin 2005 and Yin in Fields demonstrates the importance of those publications. The actual research papers are plainly at a more detailed level. But all are dependent on what the authors actually want to publish.
- I will mention briefly certain other papers relied upon by GSK: Sakurai 1999, Zhao 2000 and Sastre 2005 (from the Melero group), but I do not propose to go into the debates about what was being suggested on the basis of each of those papers. The enduring suggestion from GSK was that the authors did not recommend vaccinating with the prefusion form and/or never went on to develop a stabilised prefusion subunit vaccine, but it was plain that the authors were investigating other issues in RSV. In short, I do not consider that any of those papers shed any light on what was CGK or obvious by the time of the priority date.
Stewart-Jones 2018
- The final point relied on by GSK was the fact that it was not until 2018 that a PIV F subunit vaccine was developed. They point to the Stewart-Jones 2018 paper in which the authors propose to stabilise the PIV F in the prefusion form as a vaccine and achieve impressive results. Yin 2005 is cited (but not Yin), as well as McLellan 2013A & B and McLellan 2011.
- GSK suggested that the PIV field, in which the Yin 2005 and Yin 2006 papers lay and for which they had a crystal structure, now took the lead from the RSV field in terms of vaccine design by piggybacking on the work of McLellan. Dr Johnson agreed that 'things came full circle'. GSK suggested this is further evidence of inventiveness on the basis that Stewart-Jones 2018, despite citing Yin 2005, arrived at an approach of vaccinating by stabilising the prefusion form only once it had been seen from McLellan 2013 and not directly from the earlier structural studies.
- Stewart-Jones 2018 reports on a wide-ranging study investigation the utility of prefusion PIV 1-4 F vaccination. They engineered over 100 prefusion-stabilised PIV3 F variants and tested their efficacy by antigenic screening, examining the effects of introducing a variety of non-native disulphide bonds and cavity-filling mutations.
- I have incomplete information about what happened in the RSV field and I have almost none as regards the situation in the PIV field, or why it took so long to undertake a structure-based design of a PIV subunit vaccine. The work reported in this paper was primarily conducted by Peter Kwong's group at the NIH. There is a hint in the introduction that this work might have been prompted by Barney Graham's report a year earlier of the results of a clinical trial of an RSV prefusion F vaccine. Overall, I have so little information I decline to place any reliance on this paper.
- GSK's reliance on Stewart-Jones 2018 is the epitome of the wrong approach to secondary evidence. That paper was tab 89 (of 90) in two bundles of scientific materials, to which one can add two substantial bundles of cross-examination materials and other exhibits. Merely putting a paper in a bundle (even if it is agreed with the other side) does not begin to explain the case it is there to support, particularly when many other papers were never referred to. This example reinforces the need for a case on secondary evidence to be properly pleaded.
Dr Taylor's evidence
- GSK pointed to Dr Taylor's evidence that the earliest review article that Dr Taylor was able to identify that refers to the prefusion or post-fusion RSV F protein in the context of RSV vaccine development was Hurwitz 2011, and even then, it is the post-fusion form which is reported. GSK noted that the Hurwitz review refers to work by Swanson published in 2011 in connection with this post-fusion vaccine. Dr Taylor was otherwise not pressed on this point, indeed, on account of her limited evidence as a whole, there is very little in the evidence of Dr Taylor which goes to the issues of secondary evidence.
Dr Johnson's evidence
- During her cross-examination, Dr Johnson noted there were a limited number of labs working on protein subunits post 2003. That is, a handful versus the larger number of labs that were working on PFP or FG and doing both the primary research as well as the clinical trials. So, it was not the case that subunits were completely put to the side, it was a shift in priorities, subunit vaccines were de-prioritised. Dr Johnson noted that if one wanted to look at those inversions, almost, there would be a marked drop in the number of labs actively working on protein-based vaccine and a great expansion in the number working on vector-based or, as Dr Collins or the group from the Netherlands shared their reverse genetic system, those types of approaches.
- Implicit in GSK's case was the notion that a Skilled Team was sitting there, ready and able to act immediately on either Yin or the Jardetzky disclosures. In her evidence, Dr Johnson injected a much-needed dose of the practical realities in the RSV field:
i) First, the available funding. In comparison to the enormous resources available to the HIV field, there was only a very small pot of money in the RSV field, for which there was strong competition between the research groups. Dr Johnson suggested that there was not enough "money to build out and expand your teams". There was a specific mention of how the process of grant applications for research project work, and that a research team cannot change course suddenly - this was identified by Dr Johnson as the "major deterrent to why this was not acted on earlier". It is also worth bearing in mind Dr Johnson's evidence that, "in RSV, when you are competing for such a small pot of money you are going to guard that novelty that would get you that grant a little more closely than, say, if you were working on an HIV vaccine."
ii) Second, existing commitments. It is worth teasing out this point from Dr Johnson's answer just quoted. Having gone to the trouble of competing for grant money with a specific proposal, won the grant and embarked on implementing the proposed research, research groups would not simply drop their existing research and pick up a new opportunity immediately. The point applies with equal force to Dr Johnson, even though she was working at NIH because resources are always limited.
iii) Dr Johnson's own situation provides a good example. In this regard I refer to [373] above.
iv) More generally, Dr Johnson indicated that the inspiration to make the vaccine in the prefusion form was available in Yin, but the RSV field did not have the vaccinologist necessarily willing to take on that structural problem as a dedicated task until Jason McLellan joined the RSV field peripherally in 2010. Her point was the fact that no-one apparently took it forward does not prove that the invention was not obvious in 2007.
- So far as timing is concerned, in terms of the pre-priority case, it is unrealistic to suggest that in the 8 weeks between the Jardetzky disclosures and the Priority Date, someone could have managed to make and make public the results of work on stabilising a prefusion F RSV antigen, for use in a vaccine. Yin was published in January 2006, almost 2 years before the first priority document for the Patents, but the evidence suggested that only a fully prepared and dedicated lab with structural expertise could have stabilised a prefusion F RSV antigen in that time.
- With all the factors I have mentioned above in mind, the time taken by McLellan et al (2010-2013) and the Novartis group (from an unknown date, but one might assume 2006-July 2009) do not, in my view, support GSK's contentions.
- I believe I have assessed all the elements of GSK's case on secondary evidence (some of which cross-over into the primary evidence), but to the extent that I have omitted anything (a) it is unlikely to have any real significance and (b) I do not think it is open to GSK to complain, bearing in mind the complete lack of any statement of their case, other than in their written closing submissions.
- Drawing all these threads together and stepping back from the detail, I can now state my observations and conclusions.
- First, at an earlier stage I was sorely tempted simply to dismiss GSK's reliance on secondary evidence on the basis that it was not pleaded. However, largely because Pfizer raised no objection to GSK's pursuit of it in cross-examination, which meant that substantial evidence was led on the point, I did not consider that it would be right for me to ignore the secondary evidence.
- Second, as the authorities make clear, secondary evidence must be kept in its place. Its usefulness must depend in significant part on how complete and how testable it is. It is often key to hear oral evidence from any real-world workers relied on, and to have a robust documentary record in order to assess why the workers in question succeeded or failed. In this case, I had neither. Furthermore, when the participants appear to have had other priorities, plus restrictions caused by limited resources, it becomes even more important to have evidence from real-world participants as to what was going on.
- Third, I keep in mind the following points:
i) First, there was no evidence to support GSK's characterisation of the problem - what I referred to as their 'stated problem'. This appears to have been pure advocacy. None of the experts identified this as the problem to be overcome. There is no mention in the Patents that this was the problem to which the Patents provide the solution. It may be a reasonable inference that the previous subunit F vaccines were in the postfusion form (otherwise they would have produced much better protection) but that conclusion is merely a coincidental byproduct of the solution(s) in the Patents.
ii) Instead, the post-priority literature suggests that the problem was how to achieve sufficient stabilisation of the prefusion form to be useful as an immunogen and vaccine. On this point, my impression is that there was a marked difference between what Skilled Teams would regard as sufficient stabilisation and the seemingly minimal degree of stabilisation which is claimed in claim 1 of EP258.
iii) Indeed, far from demonstrating that those in the art remained blind to the advantages of the prefusion form, the post-priority literature indicates they were well aware that a stabilised prefusion antigen would be likely to afford better protection. See Swanson 2011.
iv) It is too simplistic, in my view, to conclude that McLellan investigated the postfusion form because his group missed the significance of the prefusion form. One has to take into account the ready availability of the stable postfusion form in comparison with the metastable prefusion form and the work required to stabilise it.
v) Finally, whilst I fully accept that sometimes even highly skilled people in the field can miss the significance of that which is contended to be obvious, I find it difficult to accept that McLellan's group or any of the other authors mentioned above had forgotten the 'basic principle' to which Dr Johnson drew attention (see [381] above).
- All these conclusions must remain tentative, precisely because the Court has a very incomplete picture and a lot of theories which are not supported by hard evidence from the participants.
- Finally, I should note that GSK's arguments are capable of being characterised as a mindset argument, particularly when I take into account one of the main thrusts of Dr Taylor's evidence. I have already noted her emphasis on Calder 2000 and her point that the monoclonal antibodies which neutralized virus infectivity and inhibited fusion bound to both forms of the F protein. Furthermore, a key point she made in her written evidence was that RSV Vaccinologists at the priority date were not addressing themselves to the question of antigenic differences between the pre- and postfusion forms of the F protein. She made the point that Yin does not address this subject. She said that it is only with hindsight that we know there are relevant antigenic differences between the two forms of the F protein.
- However, these points made by Dr Taylor were a key part of why she disagreed with the key points made by Dr Johnson in her [124] and [125], on which I continue to reserve my conclusions.
- Before I make my final conclusions, there are some further considerations I should mention.
- The first concerns the overall complexion of the case. Pfizer's case on the Skilled Team and their CGK was clearly set out in the first reports of Dr Johnson and Professor Weissenhorn. GSK's position was equally clear from Dr Taylor's reports. The principal lines of GSK's defence were (a) the composition of the Skilled Team and (b) their CGK. There was almost no dispute as to what was disclosed by the written prior art. GSK attempted to cast doubt on what Professor Jardetzky had said when speaking to his slide presentation, but I have resolved those factual issues. As far as I could detect, there was no cross-examination on the detail of the obviousness allegations (other than the general allegation of hindsight). By way of example, there was no suggestion that it was not obvious to create the prefusion F construct as a single polypeptide.
- The second concerns the import of GSK's case on secondary evidence and their related attack on [124] and [125] of Dr Johnson's first report. The final issue I must assess is this combination of factors:
i) the suggestion that these CGK points were only assembled or brought to the fore with the benefit of hindsight;
ii) that, at the time, no-one who had read Yin or attended the Jardetzky presentation joined the dots;
iii) the suggestion that 'the penny did not drop until 2013' which actually resolves into two separate points: first, that it took the highly skilled team of Jason McLellan to join the dots and, second, they did not do so until their work which was published in McLellan 2013B.
- As to these factors:
i) These CGK points were not an obscure collection of pieces of knowledge. These were basic principles to anyone addressing their minds to the prefusion and postfusion forms of RSV, PIV or other class 1 fusion viruses, a point reflected in the support for them in the textbooks. Accordingly, my interim conclusion (see [384] above) on Points A, B & D in [124] and [125] of Dr Johnson's first report becomes final.
ii) It was clear from Dr Taylor's evidence that she did not 'join the dots', but she closed her mind to anything outside RSV. Dr Johnson did 'join the dots' at the time, but chose not to follow the route, for the reasons she explained.
iii) I have no evidence from anyone in the McLellan group, or anyone else in the field.
- However, GSK's argument that the 'penny did not drop until 2013' is too simplistic, in my view. Swanson 2011 and McLellan 2011 are both inconsistent with the notion that those in the art did not realise that it would be beneficial to attempt to stop the transition from the prefusion form to the postfusion form. Swanson 2011 in particular confirms that all stages of the activation process were under consideration.
- The third point is the one I mentioned in [786] above. However, there was never any suggestion that all the antigenic sites on the F protein had been identified in Calder or anywhere else. Dr Johnson never suggested that it was certain that the stabilised prefusion form would work as a vaccine, merely that the most effective neutralizing antibodies generated in vivo would be likely to bind to the prefusion conformation of the F protein. In other words, there was a reasonable expectation of success providing sufficient motivation to investigate stabilisation of the prefusion form.
- Next are the points I left over in [668] above. However, as far as I can tell, there was no evidence that the Skilled Team would be unable to find a source of RSV prefusion protein. As for GSK's suggestion that the Skilled Team would not be able to stabilise the prefusion form either at all or without undue burden, this suggestion is, of course, directly contrary to their argument and my finding on the plausibility issue (see below). This argument was probably made (and could only be sustainable) on the basis of GSK's view of the Skilled Team (i.e. without specialist structural biology expertise).
- Professor Weissenhorn gave clear evidence that the structural biologist in the Skilled Team understood that different stabilisation strategies could be employed, a number of which had already been developed and tested in HIV-1 with the aim of preserving the native Env trimer conformation. The most common one was the addition or fusion of a small trimerization domain in place of the transmembrane domain. The structural biologist would have been aware of a number of common and well-characterized trimerization domains, such as trimeric versions of GCN4, a coiled-coil domain described by the Kim lab in the early 1990s, or the foldon domain.
- However, when assessing the secondary evidence, I noted there were indications that it did take time and effort to achieve a level of stabilisation of the prefusion form that the groups considered acceptable. As I also noted, that degree of stabilisation was likely to be well above that provided by merely adding a GCN4 trimerisation domain.
- As far as I can tell, neither Dr Taylor nor Professor Wilkinson gave any evidence to support GSK's submissions on those matters. Nor were these points put to either Dr Johnson or Professor Weissenhorn. Naturally, I will be corrected if I am wrong.
- Finally, I should consider again the suggestion from GSK that Pfizer's arguments required a super-skilled structural biologist. It is true that all the real-life teams working in this area had very impressive and wide-ranging skills and were engaged in cutting edge research. However, I have accepted almost all of Professor Weissenhorn's evidence on the CGK of the structural biologist member of the team. In the course of doing that, I satisfied myself that that knowledge was required to implement either of the Patents.
- Overall, I had clear primary evidence of obviousness from impressive expert witnesses, Dr Johnson and Professor Weissenhorn. To the extent that there was contrary evidence given by Dr Taylor and Professor Wilkinson, it was not persuasive and in particular, I have dismissed the restrictive approach taken by Dr Taylor and the inadequate way in which Professor Wilkinson was instructed. Against the prima face clear case of obviousness on the primary evidence, I did not find the secondary evidence either complete enough or anywhere near persuasive enough to displace that prima facie case. In the circumstances, I find both EP258 and EP710 invalid for obviousness over each piece of prior art.
- At [39] above, I mentioned that GSK's case on secondary evidence was developed in a very unsatisfactory manner. The case that GSK actually ran (i.e. as it emerged in their closing argument) ought, in my view, to have been pleaded so that (a) Pfizer could have investigated the matters pleaded and filed a responsive pleading, (b) appropriate case management directions could have been given, (c) the pleaded case could have been considered by the experts instructed on each side and dealt with in the written expert evidence, (d) the cross-examination could have been directed appropriately and (e) it would not have been possible for the case being run to undergo material change, without the pleaded case being amended, all of which would have enabled the case on secondary evidence to have been considered and decided in a much more focussed and straightforward manner.
- In making those observations, I do not intend to rule out the possibility that a much simpler secondary evidence case cannot be set out in an expert's report, provided that the opposing party and their expert have a proper opportunity to respond to it. That may require provision for additional evidence addressing the point, so these sorts of problems need to be anticipated. In future, however, if a party does not plead its case on secondary evidence, it will run the risk of an objection to it being upheld and/or the Court refusing to take it into account. The rules of pleading apply just as much to Patent cases as any other type of case.
- Pfizer's insufficiency case is limited to a single point that engages matters of undue burden and uncertainty, together with an enablement squeeze over the prior art.
- "Classical insufficiency" will arise where the patent does not give sufficient directions to enable a product to be made without undue burden. Kitchin J (as he then was) summarised the law in Eli Lilly and Company v Human Genome Sciences Inc [2008] EWHC 1903 (Pat) at paragraph 239:
"The specification must disclose the invention clearly and completely enough for it to be performed by a person skilled in the art. The key elements of this requirement which bear on the present case are these:
i) the first step is to identify the invention and that is to be done by reading and construing the claims;
ii) in the case of a product claim that means making or otherwise obtaining the product;
iii) in the case of a process claim, it means working the process;
iv) sufficiency of the disclosure must be assessed on the basis of the specification as a whole including the description and the claims;
v) the disclosure is aimed at the skilled person who may use his common general knowledge to supplement the information contained in the specification;
vi) the specification must be sufficient to allow the invention to be performed over the whole scope of the claim;
vii) the specification must be sufficient to allow the invention to be so performed without undue burden."
- In relation to what amounts to undue burden, I was referred to Aldous J in Mentor Corp v Hollister Inc [1991] FSR 557 at 562:
"[The skilled person] must seek success. He may need to carry out the ordinary methods of trial and error, which involve no inventive step and generally are necessary in applying the particular discovery to produce a practical result. In each case, it is a question of fact, depending on the nature of the invention, as to whether the steps needed to perform the invention are ordinary steps of trial and error which a skilled man would realise would be necessary and normal to produce a practical result."
- Aldous J went on to say that he regarded his view as being consistent with what the Technical Board of Appeal of the EPO had said in T 226/85 Unilever / stable bleaches at [8]:
"Even though a reasonable amount of trial and error is permissible when it comes to the sufficiency of disclosure in an unexplored field or - as it is in this case - where there are many technical difficulties, there must then be available adequate instructions in the specification or on the basis of common general knowledge which would lead the skilled person necessarily and directly towards success through the evaluation of initial failures or through an acceptable statistical expectation rate in case of random experiments."
- As Floyd LJ put it in Anan Kasei at [23], having referred to Kirin-Amgen:
"The House of Lords did not throw any doubt on the principle that a claim is not rendered insufficient because there is some room for doubt, or fuzziness, at the edge of the claim. The claim in Kirin-Amgen was insufficient because it was conceptually uncertain."
- Floyd LJ went on at [25] to explain that such cases arise where the process of interpretation could not resolve the question of what the patentee had in mind for the necessary test. Lewison LJ put it similarly at [101]:
"If the court cannot ascertain the boundary, having used all the interpretative tools at its disposal, it must conclude that the specification does not disclose the invention clearly enough and completely enough for it to be performed by a person skilled in the art."
- In this case Pfizer relies on the inability of the skilled team to be able without undue burden to tell whether an antigen falls within the claims as a result of the lack of teaching in the Patents and the uncertainty in scope of the term "stabilized" and how it is to be tested.
- Claim 1 of EP258 and Claims 23 and 24 of EP710 include a requirement that the trimerization domain position C-terminal to the F1 domain of the RSV F antigen "stabilizes the prefusion conformation of the F protein."
- Pfizer say that the specification is not sufficiently enabling to allow the skilled addressee to determine, without undue burden, whether an RSV F antigen is stabilized in the prefusion conformation. It is therefore convenient to note at this point the issues which Pfizer said were not dealt with in the Patent:
i) There is no teaching as to which test and conditions are used to determine stabilisation.
ii) There is no teaching as to which result would amount to stabilisation in the prefusion conformation.
iii) There are no data in the Patents which show that the RSV F antigen produced following the teaching of the Patents is in the prefusion conformation.
iv) There is no data as to its stability in such a conformation.
v) The specification therefore does not teach what result of what test, conducted under what conditions, would meet the 'stabilizes' requirement.
- GSK submitted that measuring the degree of stabilisation does not matter, because evidence from Professors Wilkinson and Weissenhorn provided there will be a level obtained with a trimerization domain on its own and if more features are added, stabilising will only go up.
- The invention is put into effect by making a construct with a heterologous trimerization domain (with or without furin cleavage sites and other modifications). The Patents teach that those modifications will stabilise the prefusion form. The amount of stabilisation is an inherent property of the construct and does not need to be measured.
- I am also satisfied that there are at least four methods which were not unduly burdensome by which the skilled team could test their construct to see whether it has adopted the prefusion form. The evidence and Patent at [0062] pointed to the following methods:
i) X-ray crystallography
ii) Electron microscopy
iii) Liposome association assays
iv) Producing monoclonal antibodies specific to the pre or post fusion conformation and determining the conformation by its binding to the antibodies.
- I have concluded that there is no need to measure the degree of stabilisation in order to work the invention, but in any event their construct could be tested. The classical insufficiency case therefore fails.
- AgrEvo has been considered in many cases in this jurisdiction and the legal principle is not controversial between the parties. AgrEvo was considered most recently by Court of Appeal in Sandoz & Teva v BMS [2023] EWCA Civ 472, citing the explanation given by Floyd LJ in Generics v Yeda [2013] EWCA Civ 925 at [39] as part of their consideration of the case law.
"39. As with any consideration of obviousness, the technical results or effects must be shared by everything falling within the claim under attack. This follows from the fundamental principle of patent law, which underpins many of the grounds of objection to validity, that the extent of the monopoly conferred by a patent must be justified by the technical contribution to the art. If some of the products covered by a claim demonstrate a particular property, but others do not, then the technical problem cannot be formulated by reference to that property. Either the products which do not exhibit the property must be excised from the claim by amendment, or the problem must be formulated by reference to some other, perhaps more mundane, technical contribution common to the whole claim."
- On the facts of this case, I do not think that AgrEvo adds anything to the grounds of obviousness and insufficiency with which I have dealt above. However, in case I was wrong in finding that the Patent involves no inventive step (i.e. if I was wrong in finding that the Patent made no contribution to the art), I should deal with Pfizer's argument that the Patent was nevertheless invalid under the AgrEvo principles as set out by Floyd LJ because its claims went beyond its alleged technical contribution. This, Pfizer argued, was because if any more stability is required to satisfy the claim, then it is outside of the plausible technical contribution of the patent.
- I reject this argument. The technical contribution of the Patents extends to the use of a PreF antigen as an immunogen to protect against RSV. The claims are limited to embodiments in which the prefusion conformation is stabilized. The skilled person knows how to stabilise the PreF antigen according to the specification. In the words of Floyd LJ, those claims are "restricted to the subject matter which makes good the technical contribution" and are not, therefore, invalid on the AgrEvo basis.
- Accordingly, if (contrary to my findings) the Patent had involved an inventive step, I would not have concluded that the Patent was nevertheless invalid on the basis of AgrEvo obviousness.
- There was not very much dispute about the applicable law. Although the Court of Appeal had handed down its judgment in Sandoz & Teva v. Bristol-Myers Squibb [2023] EWCA Civ 472 on 4 May 2023 (shortly before the Trial started, and following the decision of the Enlarged Board of Appeal in G2/21), the submissions made to me were framed in terms of the relevant principles and key cases having recently been summarised by Meade J in Sandoz & Teva v. Bristol-Myers Squibb [2022] EWHC 822 (Pat), referring to the three-step test from Fibrogen v. Akebia [2021] EWCA Civ 1279 at [53]:
i) First, what falls within the scope of the claimed class?
ii) Second, what does it mean to say that the invention works?
iii) Third, is it possible to make a reasonable prediction the invention will work with substantially everything falling within the scope of the claim?
- Reference was also made to the point made by Lord Sumption in Warner-Lambert LLC v Generics (UK) Ltd (t/a Mylan) [2018] UKSC 56 at [36], that plausibility is a "relatively undemanding" test. In summary, the specification must disclose some reason for supposing that the assertion as to the technical effect is true, i.e. something that would cause the skilled person to think that there was a reasonable prospect that the assertion would prove to be true. That may be experimental data, or it may consist of a priori reasoning.
- In the circumstances of this case, the developments in G2/21 and the analysis of that decision in the judgment of the Court of Appeal in Sandoz & Teva v. Bristol-Myers Squibb are not material to the issue here, so I do not need to lengthen this judgment further by discussing them.
- The allegation concerns whether antigens in the absence of an adjuvant are plausibly effective for preventing or treating an RSV infection or RSV associated disease.
- Pfizer's case is that the specification of the Patents only renders plausible that the RSV F antigens claimed (the constructs tested in the Examples) are effective for use to prevent or treat RSV associated diseases when administered with an adjuvant. Pfizer submit that the specification does not make any such use plausible without an adjuvant; this is so for the construct exemplified, and even more so for other (non-exemplified) antigens which could be made in accordance with the claims.
- In order to assess the allegation of plausibility, I shall apply the approach as set out in Fibrogen.
- Firstly, if the antigen is not stabilised, it does not fall within the claims. The claimed compound of relevance here is the immunogen suitable for preventing RSV infection.
- GSK contend that adjuvants are not the core of the invention and that even if it were not plausible that a vaccine could be made without an adjuvant this claim would not be invalid. They provide that the purpose of the product for each of the claims in issue is vaccinating against RSV.
- It was common general knowledge at the priority date that adjuvants enhance the immune response to the antigens with which they are combined. The evidence advanced by both Drs Johnson and Taylor explained that the vaccination of RSV-experienced individuals does not necessarily require an adjuvant, although they are beneficial (and provide more benefit to RSV naïve individuals).
- This was supported by the examples 5-7 in the patent, each of which concerned mice that were immunised twice at a fortnight's interval with PreF in various doses. Some mice were immunised with adjuvant, others without. In summary, the aims of the examples are as follows: Example 5 shows that the antigen was immunogenic (i.e., produced an antibody response); Example 6 shows that the immune response comprised neutralising antibodies; and Example 7 shows that the response was protective against disease.
- The data from Example 5 show an antibody response, both with and without an adjuvant. Examples 6 and 7 showed that a response is observed with PreF administered alone, albeit a more modest one than when used with an adjuvant. The response is shown in Example 6 to elicit neutralising antibodies and in Example 7 to be protective. These experiments were conducted in mice which were not exposed to RSV, and so are in greater need of an adjuvant to assist in mounting an immune response. The position with mice is very different to the position with RSV experienced humans. Dr Taylor noted that an "adjuvant may be less important" in the adult population.
- When asking whether a claim is plausible across its breadth, one does not look at every potential variant. Here we are talking about adjuvants. The patent is not about adjuvants, that is not the problem. It does not set up a problem about adjuvants and seek to solve that. The fact that antigen may be presented in different ways, some of which may be more efficacious than others, does not mean that the problem is not solved across the breadth of the claim.
- In light of the above, this renders plausible that PreF antigens in the presence or absence of an adjuvant are immunogenic.
- The law was not in dispute between the parties. An Arrow declaration is a declaration that a product, process or use was lacking in novelty or obvious as at the priority date of a patent application. The point of such declaration is that it is in effect a declaration that the claimant will have a Gillette defence to any subsequent claim for patent infringement in relation to that product, process or use: see Gillette Safety Razor Co v Anglo-American Trading Co Ltd (1913) 30 RPC 465.
- The key principles can be found in the judgment of Henry Carr J in Fujifilm Kyowa Kirin Biologics Co Ltd v AbbVie Biotechnology Ltd [2017] EWHC 395 (Pat) at [365]-[371]. In summary, he held that the Court must consider:
i) justice to the claimant;
ii) justice to the defendant;
iii) whether the declaration will serve a useful purpose. The attainment of commercial certainty in patent cases can constitute a useful purpose. The spin-off value of a judgment in other countries may be such a factor, but a declaration sought solely for the benefit of foreign courts will rarely be justified; and
iv) whether or not there are any other special reasons why the court should or should not grant the declaration.
- There have since been a number of decisions on Arrow declarations since the jurisdiction of the Patents Court to grant an Arrow declaration was confirmed by the Court of Appeal in Fujifilm Kyowa Kirin Biologics Co Ltd v AbbVie Biotechnology Ltd [2017] EWCA Civ 1. The jurisdictional position was summarised by Floyd LJ delivering the judgment of the Court at [98] as follows:
"... we do not consider that there is any issue of principle which prevents the granting of Arrow declarations in appropriate cases. Drawing the threads together:
(i) A declaration that a product, process or use was old or obvious at a particular date does not necessarily offend against s.74 of the Act.
(ii) Such a declaration may offend against the Act where it is a disguised attack on the validity of a granted patent.
(iii) Such declarations do not offend against the scheme of the EPC or the Act simply because the declaration is sought against the background of pending divisional applications by the counter-party.
(iv) On the other hand the existence of pending applications cannot itself be a sufficient justification for granting a declaration.
(v) Whether such a declaration is justified depends on whether a sufficient case can be made for the exercise of the court's discretion in accordance with established principles."
- Pfizer seeks an Arrow declaration that at the Priority Date/filing date it was obvious to make an RSV antigen which inter alia resembles the prefusion conformation and the use of that antigen in the treatment or prevention of RSV-associated diseases was obvious at the priority date.
- The specific features in respect of which Arrow relief is sought are:
i) The making of a soluble recombinant RSV antigen comprising an F2 domain and an F1 domain of an RSV-A or RSV-B protein, wherein a T4 fibritin "foldon" domain is positioned C-terminal to the F1 domain and wherein, when expressed, the antigen polypeptides assemble into a trimer of F2-F1 domains that resembles the prefusion conformation of the mature, processed RSV F protein (Limb 1); and
ii) The use of such an antigen in the treatment or prevention of RSV-associated diseases (Limb 2).
- GSK resists the Arrow declaration sought for the substantive reasons that the subject matter was not obvious (as set out above) and because such a declaration would not serve a useful purpose.
- I agree that the use of a fibritin foldon domain is an obvious alternative to a trimerization domain in light of the unchallenged evidence from Professor Weissenhorn that trimerization domains such as GCN4 coiled-coil or foldon domains were common and well-characterised at the Priority Date and would be obvious ones to choose. This is supported by the cross-examination of Professor Wilkinson, who agreed that GCN4 domains were a known strategy for stabilising such proteins at the priority date and that foldon domains were as known as GCN4 domains. He also agreed that as at 2007, a fibritin foldon domain would be viewed as an obvious alternative that could be used in place of a GCN4 trimerisation domain.
- As to the second feature in respect of which Pfizer seeks Arrow relief, I refer to the obviousness analysis above.
- On this issue, Pfizer argued that the declaration will serve a useful purpose because there is a clear public health interest in having Pfizer's vaccine made available in the UK. GSK's position is that it will not seek an injunction to restrain Pfizer's use of its product in relation to the maternal indication if Pfizer submits to a "commercially acceptable licence", but that it will do so in relation to the older adult population.
- GSK says that all Pfizer relies on as to why Arrow relief would serve a useful purpose is the existence of GSK's divisionals. This in my opinion does not take into account the full picture.
- Pfizer, via Mr Gilbert's evidence, has raised a number of concerns with regards to the commercial uncertainty of GSK's next steps for its divisional applications, in particular with respect to Pfizer's ability to supply its RSV vaccine product in the UK and damage its legitimate business interests in the UK (including Pfizer's ability to compete for supply contracts and Pfizer being unable to make good on their existing commitments).
- For all of the above reasons, the declaration sought would serve a useful purpose.
- For the reasons explained above, I find:
i) EP258 and EP710 are both invalid for obviousness over each piece of prior art, but neither patent is insufficient or obvious on AgrEvo grounds.
ii) I reject the allegation of lack of plausibility.
iii) Even if valid, EP258 and EP710 would not be infringed by the Pfizer product.
iv) I grant the Arrow Declaration as sought.
- Finally, I must apologise to the parties for the delay in the production of this Judgment. A good part of the delay was caused by my involvement in the Bitcoin litigation involving Dr Wright. Some was caused by the complexity of the issues which the parties left me to determine and having to return to them after attending to other judicial commitments. I do not propose to allow such a delay to occur again.
